

Genomik - GATK Einrichtung und Ausführung
 Änderungen vorschlagen
Änderungen vorschlagen
Laut dem National Human Genome Research Institute ( "NHGRI"„Genomics ist die Untersuchung aller Gene einer Person (das Genom), einschließlich der Wechselwirkungen dieser Gene miteinander und mit der Umwelt einer Person. „
Laut dem "NHGRI", "Deoxyribonukleinsäure (DNA) ist die chemische Verbindung, die die notwendigen Anweisungen enthält, um die Aktivitäten von fast allen lebenden Organismen zu entwickeln und zu leiten. DNA-Moleküle bestehen aus zwei verdrehenden, paarweise angeordneten Strängen, die oft als Doppelhelix bezeichnet werden.“ „Der gesamte DNA-Satz eines Organismus wird sein Genom genannt.“
Sequenzierung ist der Prozess der Bestimmung der genauen Reihenfolge der Basen in einem Strang der DNA. Eine der häufigsten Sequenzierungsarten, die heute verwendet werden, nennt man Sequenzierung durch Synthese. Diese Technik verwendet die Emission von fluoreszierenden Signalen, um die Grundlagen zu bestellen. Forscher können mit Hilfe der DNA-Sequenzierung nach genetischen Variationen und Mutationen suchen, die bei der Entwicklung oder dem Fortschreiten einer Krankheit eine Rolle spielen könnten, während sich eine Person noch im embryonalen Stadium befindet.
Von der Probe bis zur Variantenidentifikation, Anmerkung und Vorhersage
Genomik kann im allgemeinen zu den folgenden Schritten eingeteilt werden. Dies ist keine umfassende Liste:
-
Probenentnahme.
-
"Genom-Sequenzierung" Verwenden eines Sequenzers zum Generieren der Rohdaten.
-
Vorverarbeitung: Beispiel: "Deduplizierung" Wird verwendet "Picard".
-
Genomanalyse:
-
Wird einem Referenzgenom zugeordnet.
-
"Variante" Identifizierung und Beschriftung, die in der Regel mit GATK und ähnlichen Tools durchgeführt werden.
-
-
Integration in das Electronic Health Record-System (EHR).
-
"Bevölkerungsstratifizierung" Und Identifizierung der genetischen Variation über geografische Lage und ethnische Herkunft.
-
"Prädiktive Modelle" Verwendung von signifikanter Single-Nukleotid-Polymorphismus.
Die folgende Abbildung zeigt den Prozess von der Probenahme bis zur Variantenidentifikation, Anmerkung und Vorhersage.
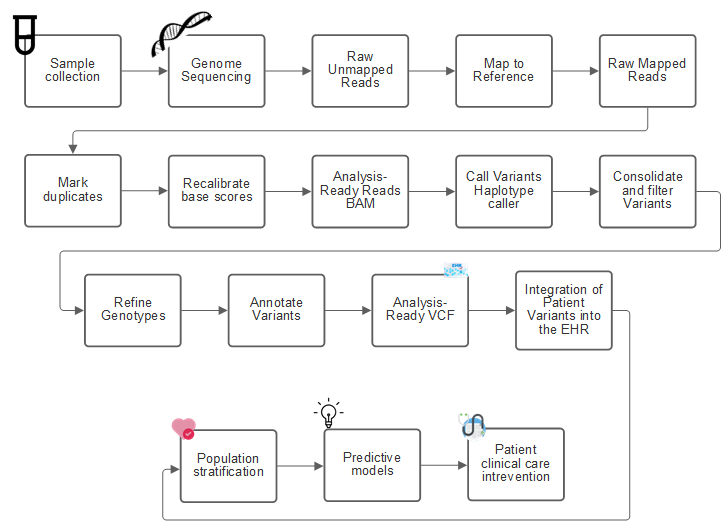
Das Human Genome Projekt wurde im April 2003 abgeschlossen und das Projekt stellte eine sehr hochwertige Simulation der menschlichen Genom-Sequenz dar, die in der Öffentlichkeit zur Verfügung stand. Das Referenzgenom initiierte eine Explosion der Forschung und Entwicklung von Genomfunktionen. Praktisch jede menschliche Krankheit hat eine Signatur in den Genen des Menschen. Bis vor kurzem nutzen Ärzte Gene, um Geburtsfehler wie Sichelzellenanämie vorherzusagen und zu bestimmen, die durch ein bestimmtes Erbmuster verursacht wird, das durch eine Änderung in einem einzelnen Gen verursacht wird. Die Schatzkammer der Daten, die das Humangenom-Projekt zur Verfügung gestellt wurde, führte zu dem Beginn des aktuellen Status der Genomfunktionen.
Die Genomik bietet zahlreiche Vorteile. Hier ein kleiner Satz von Vorteilen in den Bereichen Gesundheitswesen und Life Sciences:
-
Bessere Diagnose am Point of Care
-
Bessere Prognose
-
Präzisionsmedizin
-
Personalisierte Behandlungspläne
-
Bessere Krankheitsüberwachung
-
Verringerung unerwünschter Ereignisse
-
Besserer Zugang zu Therapien
-
Verbesserte Krankheitsüberwachung
-
Effektive Teilnahme an klinischen Studien und bessere Auswahl von Patienten für klinische Studien auf Basis von Genotypen.
Genomik ist eine "Vierköpfige," Aufgrund der Computing-Anforderungen für den gesamten Lebenszyklus eines Datensatzes, zu Erfassung, Storage, Verteilung und Analyse
Genom Analysis Toolkit (GATK)
GATK wurde als Datenwissenschaftsplattform am entwickelt "Broad Institute". GATK ist eine Reihe von Open-Source-Tools, die Genomanalysen ermöglichen, insbesondere Variantenerkennung, Identifizierung, Annotation und Genotyping. Einer der Vorteile von GATK besteht darin, dass der Satz von Tools und Befehlen zu einem kompletten Workflow gekettet werden kann. Die Hauptprobleme, mit denen sich das breite Institut befasst, sind:
-
Die Ursachen und biologischen Mechanismen von Krankheiten verstehen.
-
Identifizieren Sie therapeutische Interventionen, die auf die grundlegende Ursache einer Krankheit wirken.
-
Verstehen Sie die Sichtlinie von Varianten bis zur Funktion in der menschlichen Physiologie.
-
Standards und Richtlinien erstellen "Frameworks" Für die Darstellung von Genomdaten, Speicherung, Analysen, Sicherheit usw.
-
Standardisieren und Sozialisieren interoperabler Genom Aggregation Datenbanken (gnomAD).
-
Genom-basierte Überwachung, Diagnose und Behandlung von Patienten mit größerer Präzision.
-
Helfen Sie bei der Implementierung von Tools, mit denen Krankheiten schon lange vorhergesagt werden, bevor Symptome auftreten.
-
Schaffen und stärken Sie eine Gemeinschaft von interdisziplinären Mitarbeitern, um die schwierigsten und wichtigsten Probleme in der Biomedizin zu lösen.
Nach Angaben des GATK und des breiten Instituts sollte die Genomsequenzierung in einem Pathologielabor als Protokoll behandelt werden; jede Aufgabe ist gut dokumentiert, optimiert, reproduzierbar und konsistent über Proben und Experimente hinweg. Im Folgenden finden Sie eine Reihe von Schritten, die vom Broad Institute empfohlen werden. Weitere Informationen finden Sie im "GATK-Website".
Einrichtung von FlexPod
Für Genomik-Workloads wurde eine FlexPod Infrastrukturplattform von Grund auf neu eingerichtet. Die FlexPod Plattform ist hochverfügbar und lässt sich unabhängig skalieren, beispielsweise Netzwerk, Storage und Compute unabhängig voneinander skalieren. Wir verwendeten den folgenden Cisco Validated Design Leitfaden als Referenzarchitekturdokument zur Einrichtung der FlexPod Umgebung: "FlexPod Datacenter with VMware vSphere 7.0 and NetApp ONTAP 9.7". Sehen Sie sich die folgenden FlexPod Plattform-Highlights an:
Um die FlexPod Lab-Einrichtung durchzuführen, gehen Sie wie folgt vor:
-
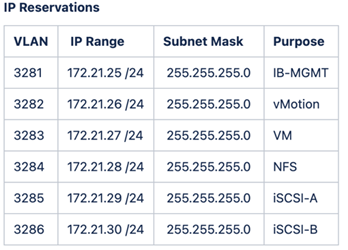
Zur Einrichtung und Validierung von FlexPod kommen die folgenden IP4-Reservierungen und -VLANs zum Einsatz.
-
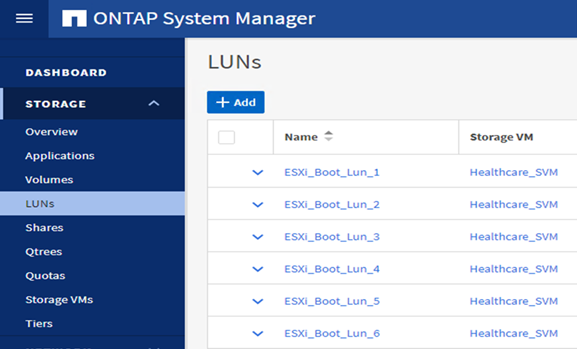
Konfigurieren Sie iSCSI-basierte Boot-LUNs auf der ONTAP SVM.
-
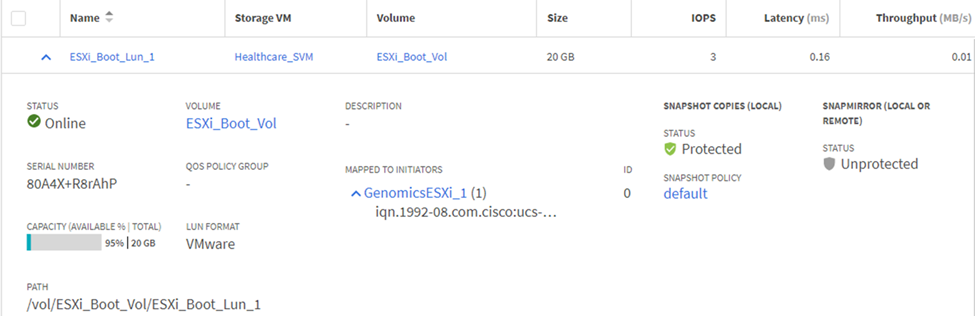
Zuordnen von LUNs zu iSCSI-Initiatorgruppen
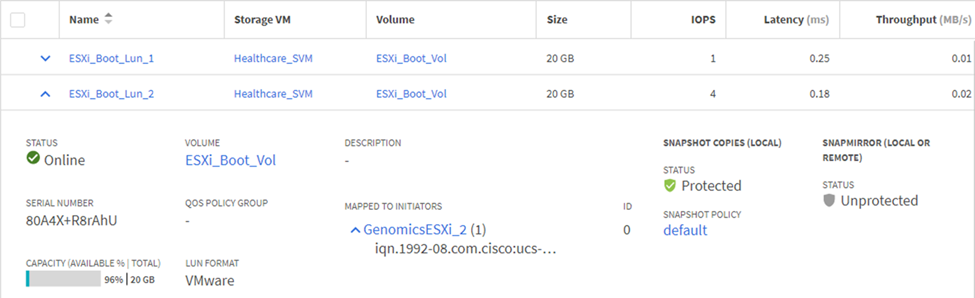
-
Installation von vSphere 7.0 mit iSCSI Boot
-
Registrieren Sie ESXi-Hosts mit vCenter.
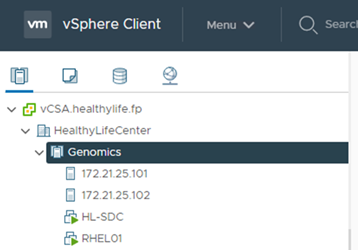
-
Bereitstellung eines NFS-Datenspeichers
infra_datastore_nfsAuf dem ONTAP Storage.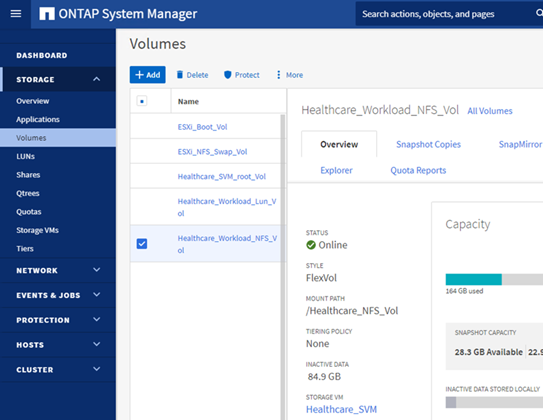
-
Fügen Sie den Datastore zum vCenter hinzu.
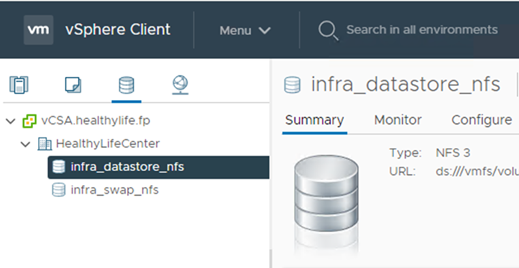
-
Fügen Sie mithilfe von vCenter einen NFS-Datenspeicher zu den ESXi Hosts hinzu.
-
Erstellen Sie mithilfe von vCenter eine VM mit Red hat Enterprise Linux (RHEL) 8.3 zur Ausführung von GATK.
-
Ein NFS-Datastore wird der VM präsentiert und bei gemountet
/mnt/genomics, Die zum Speichern von ausführbaren GATK-Dateien, Skripten, BAM-Dateien (Binary Alignment Map), Referenzdateien, Indexdateien, Wörterbuchdateien und Ausrufdateien für Variantenaufrufe verwendet wird.
GATK-Einrichtung und -Ausführung
Installieren Sie die folgenden Voraussetzungen auf der RedHat Enterprise 8.3 Linux VM:
-
Java 8 oder SDK 1.8 oder höher
-
GATK 4.2.0.0 vom Broad Institute herunterladen "GitHub-Website". Genom-Sequenzdaten werden in der Regel in Form einer Reihe von tabulatorgetrennte ASCII-Spalten gespeichert. ASCII beansprucht jedoch zu viel Platz zum Speichern. Daher wurde ein neuer Standard entwickelt, der als BAM (*.bam)-Datei bezeichnet wird. Eine BAM-Datei speichert die Sequenzdaten in komprimierter, indizierter und binärer Form. Wir "Heruntergeladen" Eine Reihe öffentlich verfügbarer BAM-Dateien für die GATK-Ausführung vom "Öffentliche Domäne". Wir haben auch Indexdateien (\*.bai), Wörterbuchdateien (*) heruntergeladen. Dict) und Referenzdatendateien (*. fasta) von der gleichen öffentlichen Domäne.
Nach dem Download verfügt das GATK-Tool-Kit über eine JAR-Datei und eine Reihe von Supportskripten.
-
gatk-package-4.2.0.0-local.jarAusführbar -
gatkSkriptdatei.
Wir haben die BAM-Dateien und die entsprechenden Index-, Wörterbuch- und Referenzgenom-Dateien für eine Familie heruntergeladen, die aus Vater-, Mutter- und Son *.bam-Dateien bestand.
Cromwell-Motor
Cromwell ist eine Open-Source-Engine, die auf wissenschaftliche Workflows ausgerichtet ist und Workflow-Management ermöglicht. Der Cromwell Motor kann in zwei laufen "Modi", Servermodus oder ein Einzelworkflowmodus. Das Verhalten des Cromwell-Motors kann mit dem gesteuert werden "Cromwell Engine-Konfigurationsdatei".
-
Servermodus. aktiviert "Rest-konforme" Ausführung von Workflows in Cromwell Engine.
-
Run-Modus. der Run-Modus eignet sich am besten zur Ausführung einzelner Workflows in Cromwell, "ref" Für einen vollständigen Satz verfügbarer Optionen im Run-Modus.
Wir nutzen die Cromwell Engine, um die Workflows und Pipelines nach Bedarf auszuführen. Die Cromwell Engine verwendet eine benutzerfreundliche "Sprache für die Workflow-Beschreibung" (WDL)-basierte Skriptsprache. Cromwell unterstützt auch einen zweiten Workflow-Skriptstandard, der als Common Workflow Language (CWL) bezeichnet wird. In diesem technischen Bericht wurde WDL verwendet. WDL wurde ursprünglich vom Broad Institute for Genome Analysis Pipelines entwickelt. Mithilfe der WDL-Workflows können verschiedene Strategien implementiert werden, darunter:
-
Linear Chaining. wie der Name schon sagt, wird die Ausgabe von Task#1 als Eingabe an Task #2 gesendet.
-
Multi-in/out. Dies ist ähnlich wie bei linearer Verkettung, da jede Aufgabe mehrere Ausgänge als Eingang zu nachfolgenden Aufgaben haben kann.
-
Scatter-Gather. Dies ist eine der leistungsstärksten EAI-Strategien (Enterprise Application Integration), die zur Verfügung stehen, insbesondere bei ereignisgesteuerter Architektur. Jede Aufgabe wird entkoppelt ausgeführt, und die Ausgabe für jede Aufgabe wird in die Endausgabe konsolidiert.
Es gibt drei Schritte, wenn WDL zum Ausführen von GATK im Standalone-Modus verwendet wird:
-
Syntax validieren mit
womtool.jar.[root@genomics1 ~]# java -jar womtool.jar validate ghplo.wdl
-
Eingabe JSON generieren.
[root@genomics1 ~]# java -jar womtool.jar inputs ghplo.wdl > ghplo.json
-
Führen Sie den Workflow mit der Cromwell Engine und aus
Cromwell.jar.[root@genomics1 ~]# java -jar cromwell.jar run ghplo.wdl –-inputs ghplo.json
Das GATK kann mit mehreren Methoden ausgeführt werden; dieses Dokument untersucht drei dieser Methoden.
Ausführung von GATK mit der JAR-Datei
Schauen wir uns eine einzelne Variante Call Pipeline-Ausführung unter Verwendung des haplotypype Variant Caller an.
[root@genomics1 ~]# java -Dsamjdk.use_async_io_read_samtools=false \ -Dsamjdk.use_async_io_write_samtools=true \ -Dsamjdk.use_async_io_write_tribble=false \ -Dsamjdk.compression_level=2 \ -jar /mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar \ HaplotypeCaller \ --input /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ --output workshop_1906_2-germline_bams_father.validation.vcf \ --reference /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta
Bei dieser Methode der Ausführung verwenden wir die lokale GATK-Ausführungs-JAR-Datei, wir verwenden einen einzigen java-Befehl, um die JAR-Datei aufzurufen, und wir übergeben mehrere Parameter an den Befehl.
-
Dieser Parameter gibt an, dass wir den aufrufen
HaplotypeCallerVariant Caller Pipeline. -
-- inputGibt die Eingabe-BAM-Datei an. -
--outputGibt die Variant-Ausgabedatei im Variantenaufrufformat (*.vcf) an ("ref"). -
Mit dem
--referenceParameter, geben wir ein Referenzgenom weiter.
Nach der Ausführung sind die Ausgabendetails im Abschnitt zu finden "Ausgabe zur Ausführung des GATK unter Verwendung der JAR-Datei."
Ausführung von GATK mit ./gatk-Skript
Das GATK-Werkzeugkit kann mit dem ausgeführt werden ./gatk Skript: Untersuchen wir nun den folgenden Befehl:
[root@genomics1 execution]# ./gatk \ --java-options "-Xmx4G" \ HaplotypeCaller \ -I /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ -R /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta \ -O /mnt/genomics/GATK/TEST\ DATA/variants.vcf
Wir übergeben mehrere Parameter an den Befehl.
-
Dieser Parameter gibt an, dass wir den aufrufen
HaplotypeCallerVariant Caller Pipeline. -
-IGibt die Eingabe-BAM-Datei an. -
-OGibt die Variant-Ausgabedatei im Variantenaufrufformat (*.vcf) an ("ref"). -
Mit dem
-RParameter, geben wir ein Referenzgenom weiter.
Nach der Ausführung sind die Ausgabendetails im Abschnitt zu finden
Ausführung von GATK mit Cromwell Engine
Wir verwenden die Cromwell-Engine, um die Ausführung des GATK zu verwalten. Schauen wir uns die Kommandozeile und ihre Parameter an.
[root@genomics1 genomics]# java -jar cromwell-65.jar \ run /mnt/genomics/GATK/seq/ghplo.wdl \ --inputs /mnt/genomics/GATK/seq/ghplo.json
Hier rufen wir den Java-Befehl auf, indem wir den übergeben -jar Parameter, um anzugeben, dass wir eine JAR-Datei ausführen möchten, z. B. Cromwell-65.jar. Der nächste Parameter wurde übergeben (run) Zeigt an, dass die Cromwell-Engine im Run-Modus läuft, die andere mögliche Option ist der Server-Modus. Der nächste Parameter lautet *.wdl Dass der Run-Modus zum Ausführen der Pipelines verwendet werden soll. Der nächste Parameter ist der Satz von Eingabeparametern für die ausgeführten Workflows.
Hier ist der Inhalt der ghplo.wdl Datei wie folgt aussehen:
[root@genomics1 seq]# cat ghplo.wdl
workflow helloHaplotypeCaller {
call haplotypeCaller
}
task haplotypeCaller {
File GATK
File RefFasta
File RefIndex
File RefDict
String sampleName
File inputBAM
File bamIndex
command {
java -jar ${GATK} \
HaplotypeCaller \
-R ${RefFasta} \
-I ${inputBAM} \
-O ${sampleName}.raw.indels.snps.vcf
}
output {
File rawVCF = "${sampleName}.raw.indels.snps.vcf"
}
}
[root@genomics1 seq]#
Hier ist die entsprechende JSON-Datei mit den Eingaben zur Cromwell Engine.
[root@genomics1 seq]# cat ghplo.json
{
"helloHaplotypeCaller.haplotypeCaller.GATK": "/mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar",
"helloHaplotypeCaller.haplotypeCaller.RefFasta": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta",
"helloHaplotypeCaller.haplotypeCaller.RefIndex": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta.fai",
"helloHaplotypeCaller.haplotypeCaller.RefDict": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.dict",
"helloHaplotypeCaller.haplotypeCaller.sampleName": "fatherbam",
"helloHaplotypeCaller.haplotypeCaller.inputBAM": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bam",
"helloHaplotypeCaller.haplotypeCaller.bamIndex": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bai"
}
[root@genomics1 seq]#
Bitte beachten Sie, dass Cromwell für die Ausführung eine in-Memory-Datenbank verwendet. Nach der Ausführung ist das Ausgabungsprotokoll im Abschnitt zu sehen "Ausgabe zur Ausführung von GATK mit Cromwell Engine."
Eine umfassende Reihe von Schritten zur Ausführung des GATK finden Sie im "GATK-Dokumentation".