

Genómica - instalación y ejecución de GATK
 Sugerir cambios
Sugerir cambios
Según el Instituto Nacional de Investigación del Genoma humano ( "NHGRI"), “la genómica es el estudio de todos los genes de una persona (el genoma), incluyendo las interacciones de estos genes entre sí y con el medio ambiente de una persona. ”
Según la "NHGRI", “El ácido Deoxirribonucleico (ADN) es el compuesto químico que contiene las instrucciones necesarias para desarrollar y dirigir las actividades de casi todos los organismos vivos. Las moléculas de ADN están hechas de dos filamentos de torsión, emparejados, a menudo llamados como una hélice doble". “El conjunto completo de ADN de un organismo se llama su genoma”.
La secuenciación es el proceso de determinar el orden exacto de las bases en una cadena de ADN. Uno de los tipos más comunes de secuenciación utilizados hoy se llama secuenciación por síntesis. Esta técnica usa la emisión de señales fluorescentes para ordenar las bases. Los investigadores pueden usar la secuenciación del ADN para buscar variaciones genéticas y cualquier mutación que pueda desempeñar un papel en el desarrollo o progresión de una enfermedad mientras una persona todavía está en la etapa embrionaria.
De muestra a identificación de variante, anotación y predicción
En líneas generales, la genómica se puede clasificar en los siguientes pasos. Esta no es una lista exhaustiva:
-
Recogida de muestras.
-
"Secuenciación genómica" uso de un secuenciador para generar los datos sin procesar.
-
Preprocesamiento. Por ejemplo: "deduplicación" uso "Picard".
-
Análisis genómico.
-
Asignación a un genoma de referencia.
-
"Variante" La identificación y la anotación se realizan normalmente con GATK y herramientas similares.
-
-
Integración en el sistema de historiales médicos electrónicos (EHR).
-
"Estratificación poblacional" e identificación de la variación genética a través de la ubicación geográfica y el origen étnico.
-
"Modelos predictivos" utilizando un polimorfismo significativo de un solo nucleótido.
En la siguiente figura se muestra el proceso desde el muestreo hasta la identificación de variantes, la anotación y la predicción.
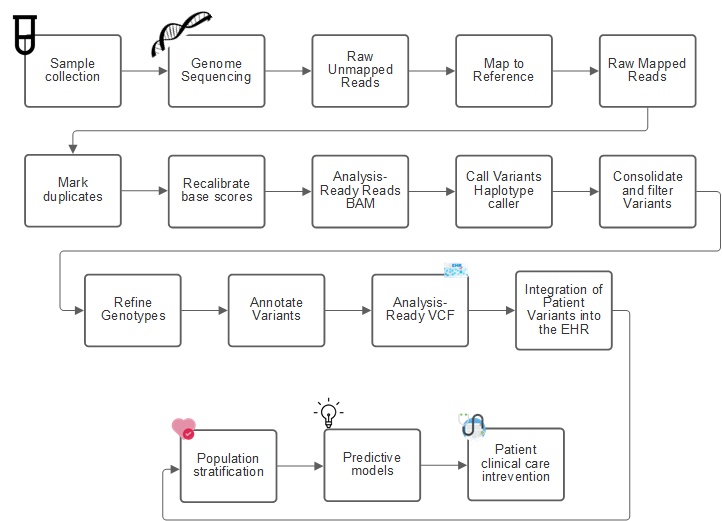
El proyecto Genoma humano se completó en abril de 2003 y el proyecto hizo una simulación de muy alta calidad de la secuencia del genoma humano disponible en el dominio público. Este genoma de referencia inició una explosión en la investigación y el desarrollo de capacidades de genómica. Prácticamente cada dolencia humana tiene una firma en los genes de ese ser humano. Hasta hace poco, los médicos estaban aprovechando los genes para predecir y determinar defectos de nacimiento como la anemia drepanocítica, que es causada por un cierto patrón de herencia causado por un cambio en un solo gen. El tesoro de los datos que el proyecto del genoma humano ha puesto a su disposición llevó a la llegada del estado actual de las capacidades genómicas.
La genómica ofrece un amplio conjunto de ventajas. A continuación se presenta un pequeño conjunto de beneficios en los ámbitos de la salud y las ciencias de la vida:
-
Mejor diagnóstico en el punto de atención
-
Mejor pronóstico
-
Medicina de precisión
-
Planes de tratamiento personalizados
-
Mejor control de enfermedades
-
Reducción de acontecimientos adversos
-
Mejora del acceso a las terapias
-
Mejor control de enfermedades
-
Participación efectiva en ensayos clínicos y mejor selección de pacientes para ensayos clínicos basados en genotipos.
La genómica es un "bestia con cuatro cabezas," debido a las demandas computacionales que se encuentran a lo largo del ciclo de vida de un conjunto de datos: adquisición, almacenamiento, distribución y análisis.
Kit de herramientas de análisis de genoma (GATK)
GATK fue desarrollado como una plataforma de ciencia de datos en el "Instituto amplio". GATK es un conjunto de herramientas de código abierto que permiten el análisis del genoma, específicamente el descubrimiento de variantes, la identificación, la anotación y el genotipado. Una de las ventajas de GATK es que el conjunto de herramientas y comandos se pueden encadenar para formar un flujo de trabajo completo. Los principales desafíos que enfrenta el instituto general son los siguientes:
-
Entender las causas profundas y los mecanismos biológicos de las enfermedades.
-
Identificar las intervenciones terapéuticas que actúen en la causa fundamental de una enfermedad.
-
Entender la línea de visión de las variantes a la función en la fisiología humana.
-
Crear estándares y políticas "marcos de trabajo" para representación de datos genómicos, almacenamiento, análisis, seguridad, etc.
-
Estandarizar y socializar las bases de datos de agregación de genomas interoperables (gnomAD).
-
Monitoreo, diagnóstico y tratamiento basados en el genoma de pacientes con mayor precisión.
-
Ayudar a implementar herramientas que predicen enfermedades mucho antes de que aparezcan los síntomas.
-
Crear y empoderar a una comunidad de colaboradores interdisciplinarios para ayudar a enfrentar los problemas más difíciles e importantes de la biomedicina.
Según GATK y el instituto general, la secuenciación del genoma debe ser tratada como un protocolo en un laboratorio de patología; cada tarea está bien documentada, optimizada, reproducible y consistente entre muestras y experimentos. El siguiente es un conjunto de pasos recomendados por el Instituto amplio, para más información, ver el "Sitio web de GATK".
Configuración de FlexPod
La validación de cargas de trabajo genómica incluye una configuración desde cero de una plataforma de infraestructura FlexPod. La plataforma FlexPod tiene una alta disponibilidad y se puede escalar de forma independiente; por ejemplo, red, almacenamiento y recursos informáticos se pueden escalar de forma independiente. Utilizamos la siguiente guía de diseño validado por Cisco como documento de arquitectura de referencia para configurar el entorno FlexPod: "FlexPod Datacenter con VMware vSphere 7.0 y ONTAP 9.7 de NetApp". Consulte los aspectos destacados de la siguiente configuración de la plataforma FlexPod:
Para realizar la configuración de FlexPod Lab, realice los siguientes pasos:
-
La configuración y validación de FlexPod Lab utiliza las siguientes reservas IP4 y VLAN.
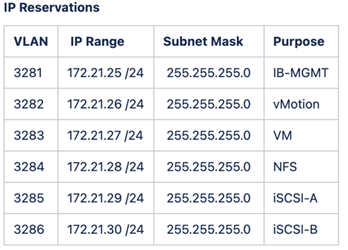
-
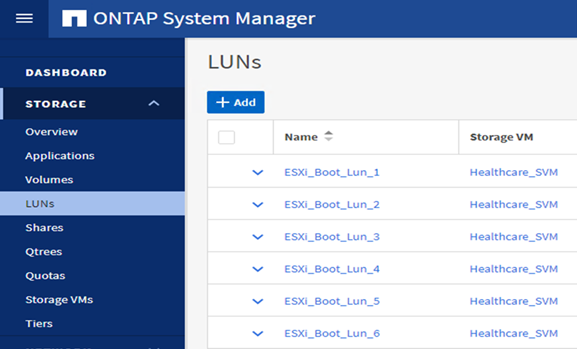
Configure los LUN de arranque basados en iSCSI en la SVM ONTAP.
-
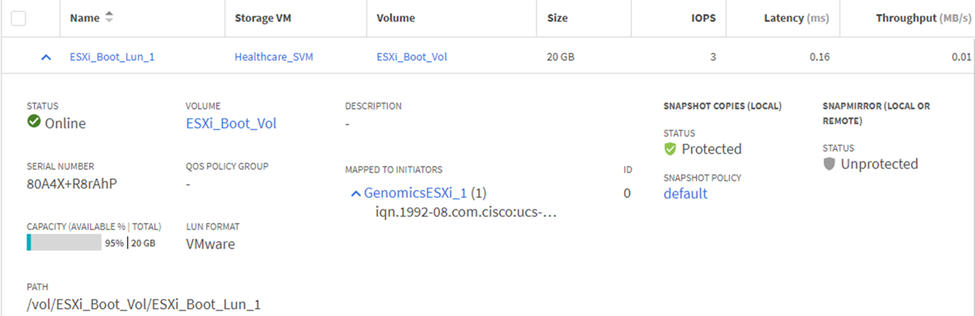
Asigne LUN a iGroups iSCSI.
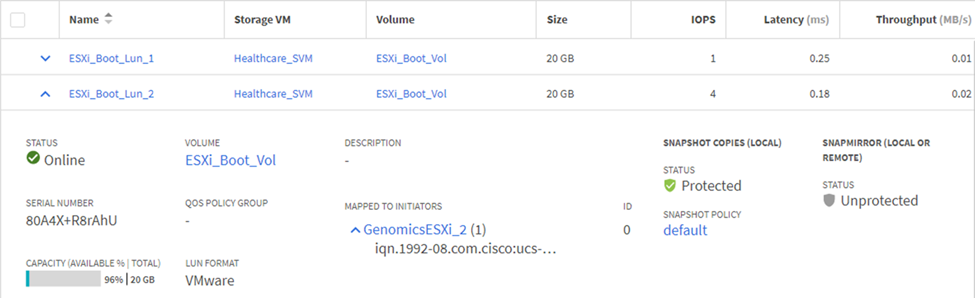
-
Instale vSphere 7.0 con arranque iSCSI.
-
Registre hosts ESXi en vCenter.
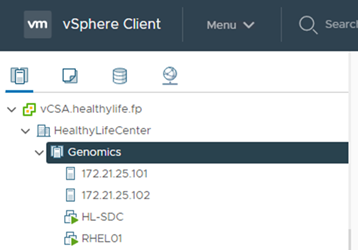
-
Aprovisione un almacén de datos NFS
infra_datastore_nfsEn el almacenamiento de ONTAP.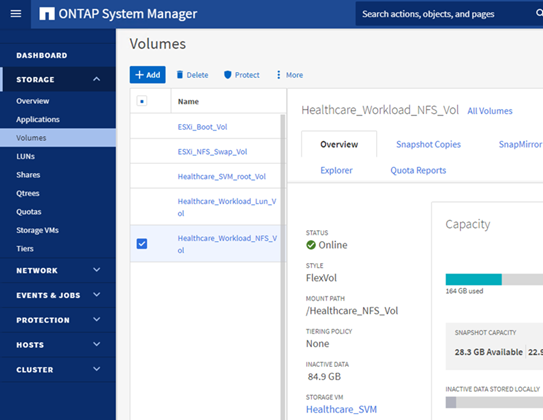
-
Añada el almacén de datos a vCenter.
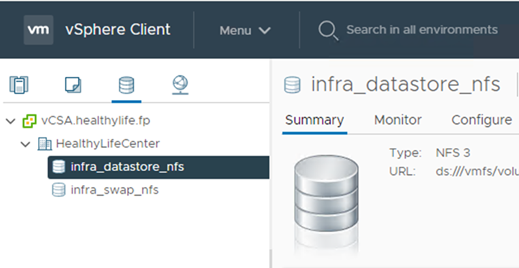
-
Con vCenter, añada un almacén de datos NFS a los hosts ESXi.
-
Mediante el para vCenter, cree un equipo virtual Red Hat Enterprise Linux (RHEL) 8.3 para ejecutar GATK.
-
Un almacén de datos NFS se presenta a la máquina virtual y se monta en
/mnt/genomics, Que se utiliza para almacenar ejecutables de GATK, secuencias de comandos, archivos de mapa de alineación binaria (BAM), archivos de referencia, archivos de índice, archivos de diccionario y archivos de salida para llamadas de variante.
Configuración y ejecución de GATK
Instale los siguientes requisitos previos en Red Hat Enterprise 8.3 Linux VM:
-
Java 8 o SDK 1.8 o posterior
-
Descargue GATK 4.2.0.0 de The Broad Institute "El sitio de GitHub". Los datos de la secuencia del genoma se almacenan generalmente en forma de una serie de columnas ASCII delimitadas por tabulaciones. Sin embargo, ASCII toma demasiado espacio para almacenar. Por lo tanto, un nuevo estándar evolucionó llamado un archivo BAM (*.bam). Un archivo BAM almacena los datos de secuencia en forma comprimida, indizada y binaria. Nosotros "descargado" Un conjunto de archivos BAM disponibles públicamente para la ejecución de GATK desde el "dominio público". También descargamos archivos de índice (\*.bai), archivos de diccionario (*. dict) y archivos de datos de referencia (*. fasta) del mismo dominio público.
Después de la descarga, el kit de herramientas GATK tiene un archivo JAR y un conjunto de secuencias de comandos de soporte.
-
gatk-package-4.2.0.0-local.jarejecutable -
gatkarchivo de script.
Descargamos los archivos BAM y los correspondientes archivos de índice, diccionario y genoma de referencia para una familia que consistía en archivos padre, madre e hijo *.bam.
Motor Cromwell
Cromwell es un motor de código abierto orientado a flujos de trabajo científicos que permite la gestión del flujo de trabajo. El motor Cromwell puede funcionar en dos "modos", Modo servidor o modo de ejecución de un único flujo de trabajo. El comportamiento del motor Cromwell se puede controlar mediante el "Archivo de configuración del motor Cromwell".
-
Modo servidor. activado "Tranquilo" Ejecución de flujos de trabajo en motor Cromwell.
-
Modo de ejecución. el modo de ejecución es más adecuado para ejecutar flujos de trabajo individuales en Cromwell, "ref" Para obtener un conjunto completo de opciones disponibles en el modo de ejecución.
Utilizamos el motor Cromwell para ejecutar los flujos de trabajo y las tuberías a escala. El motor Cromwell utiliza un motor fácil de usar "idioma de descripción del flujo de trabajo" Lenguaje de secuencias de comandos basado en (WDL). Cromwell también admite un segundo estándar de secuencias de comandos de flujo de trabajo denominado Common Workflow Language (CWL). A lo largo de este informe técnico, utilizamos el programa WDL. El WDL fue originalmente desarrollado por el Instituto amplio para oleoductos de análisis de genoma. Los flujos de trabajo de WDL se pueden implementar utilizando varias estrategias, incluidas las siguientes:
-
Encadenamiento lineal. como su nombre indica, la salida de la tarea#1 se envía a la tarea #2 como entrada.
-
Multi-in/out. esto es similar al encadenamiento lineal en que cada tarea puede tener múltiples salidas enviadas como entrada a tareas posteriores.
-
Scatter-gather. esta es una de las estrategias de integración de aplicaciones empresariales (EAI) más potentes disponible, especialmente cuando se utiliza en arquitectura basada en eventos. Cada tarea se ejecuta de forma desacoplada y el resultado de cada tarea se consolida en el resultado final.
Hay tres pasos cuando se utiliza WDL para ejecutar GATK en modo independiente:
-
Validar la sintaxis con
womtool.jar.[root@genomics1 ~]# java -jar womtool.jar validate ghplo.wdl
-
Generar entradas JSON.
[root@genomics1 ~]# java -jar womtool.jar inputs ghplo.wdl > ghplo.json
-
Ejecute el flujo de trabajo con el motor Cromwell y.
Cromwell.jar.[root@genomics1 ~]# java -jar cromwell.jar run ghplo.wdl –-inputs ghplo.json
El GATK se puede ejecutar utilizando varios métodos; este documento explora tres de estos métodos.
Ejecución de GATK usando el archivo JAR
Veamos una sola versión de la ejecución de la canalización de llamadas usando la variante de haplotipo que llama.
[root@genomics1 ~]# java -Dsamjdk.use_async_io_read_samtools=false \ -Dsamjdk.use_async_io_write_samtools=true \ -Dsamjdk.use_async_io_write_tribble=false \ -Dsamjdk.compression_level=2 \ -jar /mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar \ HaplotypeCaller \ --input /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ --output workshop_1906_2-germline_bams_father.validation.vcf \ --reference /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta
En este método de ejecución, utilizamos el archivo JAR de ejecución local GATK, utilizamos un solo comando java para invocar el archivo JAR y pasamos varios parámetros al comando.
-
Este parámetro indica que estamos invocando el
HaplotypeCallercanalización de llamada variante. -
-- inputEspecifica el archivo BAM de entrada. -
--outputespecifica el archivo de salida de variante en formato de llamada variante (*.vcf) ("ref"). -
Con la
--referenceparámetro, estamos pasando un genoma de referencia.
Una vez ejecutada, los detalles de salida se pueden encontrar en la sección "Salida para la ejecución de GATK utilizando el archivo JAR."
Ejecución de GATK usando el script ./gatk
El kit de herramientas de GATK se puede ejecutar usando el ./gatk guión. Examinemos el siguiente comando:
[root@genomics1 execution]# ./gatk \ --java-options "-Xmx4G" \ HaplotypeCaller \ -I /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ -R /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta \ -O /mnt/genomics/GATK/TEST\ DATA/variants.vcf
Pasamos varios parámetros al comando.
-
Este parámetro indica que estamos invocando el
HaplotypeCallercanalización de llamada variante. -
-IEspecifica el archivo BAM de entrada. -
-Oespecifica el archivo de salida de variante en formato de llamada variante (*.vcf) ("ref"). -
Con la
-Rparámetro, estamos pasando un genoma de referencia.
Una vez ejecutada, los detalles de salida se pueden encontrar en la sección
Ejecución del GATK con el motor de Cromwell
Utilizamos el motor Cromwell para gestionar la ejecución de GATK. Examinemos la línea de comandos y sus parámetros.
[root@genomics1 genomics]# java -jar cromwell-65.jar \ run /mnt/genomics/GATK/seq/ghplo.wdl \ --inputs /mnt/genomics/GATK/seq/ghplo.json
Aquí, invocamos el comando Java pasando el -jar parámetro para indicar que queremos ejecutar un archivo jar, por ejemplo, Cromwell-65.jar. Se ha pasado el siguiente parámetro (run) Indica que el motor Cromwell está funcionando en modo de funcionamiento, la otra opción posible es modo de servidor. El siguiente parámetro es *.wdl Que el modo Run debe utilizar para ejecutar las tuberías. El siguiente parámetro es el conjunto de parámetros de entrada de los flujos de trabajo que se están ejecutando.
Esto es lo que el contenido del ghplo.wdl aspecto del archivo:
[root@genomics1 seq]# cat ghplo.wdl
workflow helloHaplotypeCaller {
call haplotypeCaller
}
task haplotypeCaller {
File GATK
File RefFasta
File RefIndex
File RefDict
String sampleName
File inputBAM
File bamIndex
command {
java -jar ${GATK} \
HaplotypeCaller \
-R ${RefFasta} \
-I ${inputBAM} \
-O ${sampleName}.raw.indels.snps.vcf
}
output {
File rawVCF = "${sampleName}.raw.indels.snps.vcf"
}
}
[root@genomics1 seq]#
Aquí está el archivo JSON correspondiente con las entradas del motor Cromwell.
[root@genomics1 seq]# cat ghplo.json
{
"helloHaplotypeCaller.haplotypeCaller.GATK": "/mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar",
"helloHaplotypeCaller.haplotypeCaller.RefFasta": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta",
"helloHaplotypeCaller.haplotypeCaller.RefIndex": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta.fai",
"helloHaplotypeCaller.haplotypeCaller.RefDict": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.dict",
"helloHaplotypeCaller.haplotypeCaller.sampleName": "fatherbam",
"helloHaplotypeCaller.haplotypeCaller.inputBAM": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bam",
"helloHaplotypeCaller.haplotypeCaller.bamIndex": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bai"
}
[root@genomics1 seq]#
Tenga en cuenta que Cromwell utiliza una base de datos en memoria para la ejecución. Una vez ejecutado, el registro de salida se puede ver en la sección "Salida para la ejecución del GATK utilizando el motor Cromwell."
Para ver un conjunto completo de pasos sobre cómo ejecutar GATK, consulte "Documentación de GATK".