

Génomique - configuration et exécution de la GATK
 Suggérer des modifications
Suggérer des modifications
Selon l'Institut national de recherche sur le génome humain ( "NHGRI"), « la génomique est l’étude de tous les gènes d’une personne (le génome), y compris les interactions entre ces gènes et avec l’environnement d’une personne. ”
Selon le "NHGRI", "L'acide désoxyribonucléique (ADN) est le composé chimique qui contient les instructions nécessaires pour développer et diriger les activités de presque tous les organismes vivants. Les molécules d’ADN sont faites de deux torons couplés, souvent appelés « hélice double ». « L’ensemble complet d’ADN d’un organisme est appelé son génome. »
Le séquençage est le processus de détermination de l'ordre exact des bases dans un brin d'ADN. L'un des types de séquençage les plus courants utilisés aujourd'hui est appelé séquençage par synthèse. Cette technique utilise l'émission de signaux fluorescents pour commander les bases. Les chercheurs peuvent utiliser le séquençage de l'ADN pour rechercher des variations génétiques et des mutations susceptibles de jouer un rôle dans le développement ou la progression d'une maladie alors qu'une personne est encore au stade embryonnaire.
De l'identification de l'échantillon à la variante, de l'annotation et de la prédiction
À un niveau élevé, la génomique peut être classée comme suit. Cette liste n'est pas exhaustive :
-
Prélèvement d'échantillons.
-
"Séquençage du génome" utilisation d'un séquenceur pour générer les données brutes.
-
Prétraitement. Par exemple : "déduplication" à l'aide de "Picard".
-
Analyse génomique.
-
Mappage sur un génome de référence.
-
"Variante" Identification et annotation effectuées généralement à l'aide de la GATK et d'outils similaires.
-
-
Intégration au système de dossier médical électronique (EHR).
-
"Stratification de la population" et l'identification de la variation génétique entre la localisation géographique et le milieu ethnique.
-
"Modèles prédictifs" utilisant un polymorphisme significatif de nucléotide unique.
La figure suivante montre le processus de l'échantillonnage à l'identification de variante, à l'annotation et à la prédiction.
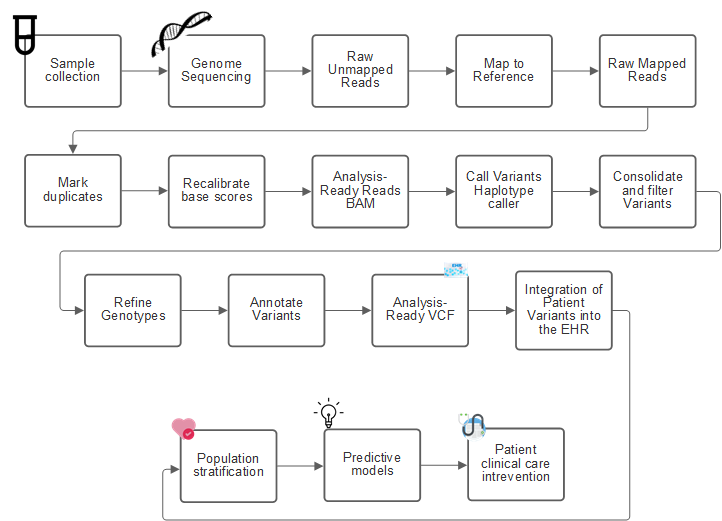
Le projet sur le génome humain a été achevé en avril 2003 et le projet a réalisé une simulation de très haute qualité de la séquence du génome humain disponible dans le domaine public. Ce génome de référence a initié une explosion de la recherche et du développement de capacités en génomique. Pratiquement chaque affection humaine a une signature dans les gènes de cet homme. Jusqu'à récemment, les médecins utilisaient les gènes pour prédire et déterminer les anomalies congénitales comme l'anémie falciforme, qui est causée par un certain modèle d'héritage causé par un changement dans un gène unique. Le Trésor des données mises à disposition par le projet du génome humain a conduit à l'avènement de l'état actuel des capacités en génomique.
La génomique offre de nombreux avantages. Voici quelques avantages dans le domaine de la santé et des sciences de la vie :
-
Un meilleur diagnostic sur le lieu des soins
-
Meilleur pronostic
-
Médecine de précision
-
Plans de traitement personnalisés
-
Une meilleure surveillance des maladies
-
Réduction des événements indésirables
-
Amélioration de l'accès aux thérapies
-
Amélioration de la surveillance des maladies
-
Participation efficace aux essais cliniques et meilleure sélection des patients pour les essais cliniques basés sur les génotypes.
La génomique est une "bête à quatre têtes," compte tenu des besoins de calcul tout au long du cycle de vie d'un dataset : acquisition, stockage, distribution et analyse.
Boîte à outils d'analyse génomique (GATK)
La GATK a été développée comme plate-forme de science des données à l' "Institut étendu". GATK est un ensemble d'outils open source qui permettent l'analyse du génome, en particulier la découverte de variantes, l'identification, l'annotation et le génotypage. L'un des avantages de GATK est que l'ensemble d'outils et/ou de commandes peut être enchaîné pour former un workflow complet. Voici les principaux défis que le large institut doit relever :
-
Comprendre les causes profondes et les mécanismes biologiques des maladies.
-
Identifier les interventions thérapeutiques qui agissent à la cause fondamentale d'une maladie.
-
Comprendre la ligne visuelle des variantes au fonctionnement dans la physiologie humaine.
-
Création de normes et de règles "frameworks" pour la représentation, le stockage, l'analyse, la sécurité, etc. des données génomiques.
-
Normaliser et socialiser les bases de données d'agrégation de génome interopérables (gnomAD).
-
Surveillance, diagnostic et traitement basés sur des génomes pour le compte de patients avec plus de précision.
-
Aider à mettre en œuvre des outils qui prédisent les maladies bien avant que les symptômes apparaissent.
-
Créer et donner les moyens à une communauté de collaborateurs transdisciplinaires pour aider à s'attaquer aux problèmes les plus difficiles et les plus importants de la biomédecine.
Selon la GATK et le large institute, le séquençage du génome doit être considéré comme un protocole dans un laboratoire de pathologie. Chaque tâche est bien documentée, optimisée, reproductible et cohérente dans l'ensemble des échantillons et des expériences. Voici un ensemble de mesures recommandées par le large Institute pour plus d'informations, voir "Site web de GATK".
Définition FlexPod
La validation d'un workload génomique inclut une configuration complète d'une plateforme d'infrastructure FlexPod. La plateforme FlexPod est extrêmement disponible et peut évoluer indépendamment. Ainsi, vous pouvez faire évoluer indépendamment le réseau, le stockage et les ressources de calcul, par exemple. Nous avons utilisé le guide de conception validée Cisco suivant comme document d'architecture de référence pour configurer l'environnement FlexPod : "FlexPod Datacenter avec VMware vSphere 7.0 et NetApp ONTAP 9.7". Découvrez les points forts de la configuration de la plateforme FlexPod suivante :
Pour effectuer la configuration du laboratoire FlexPod, procédez comme suit :
-
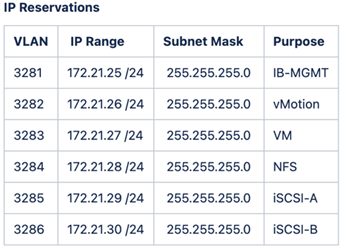
La configuration et la validation du laboratoire FlexPod utilisent les réservations IP4 et les VLAN suivants.
-
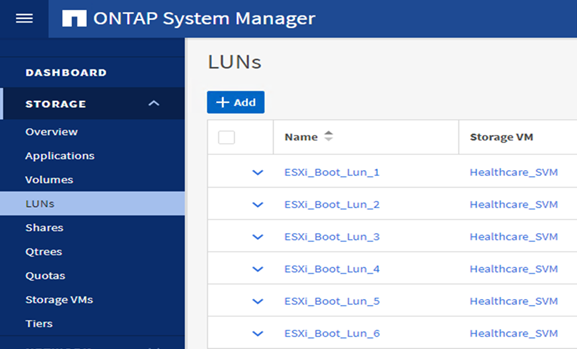
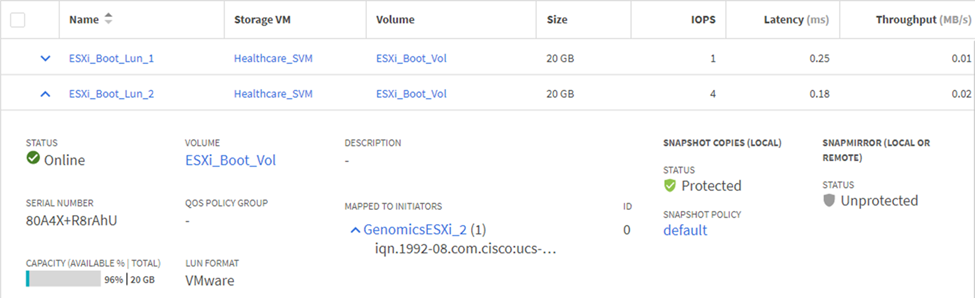
Configuration des LUN de démarrage iSCSI sur le SVM ONTAP
-
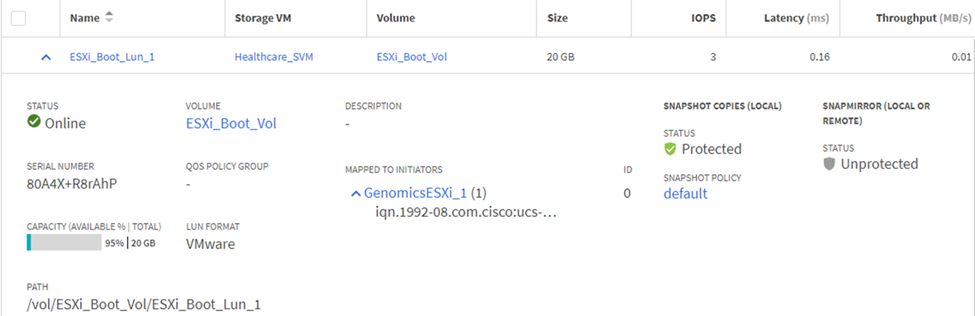
Mapper les LUN sur des groupes initiateurs iSCSI.
-
Installez vSphere 7.0 avec le démarrage iSCSI.
-
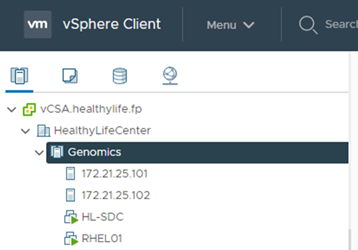
Enregistrez les hôtes ESXi avec vCenter.
-
Provisionner un datastore NFS
infra_datastore_nfsSur le stockage ONTAP.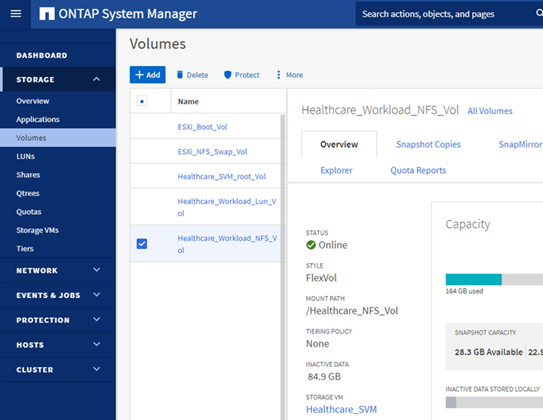
-
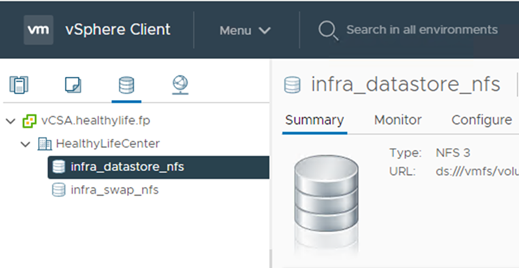
Ajoutez le datastore au vCenter.
-
À l'aide de vCenter, ajoutez un datastore NFS aux hôtes ESXi.
-
A l'aide de vCenter, créez une machine virtuelle Red Hat Enterprise Linux (RHEL) 8.3 pour exécuter GATK.
-
Un datastore NFS est présenté à la machine virtuelle et monté sur
/mnt/genomics, Qui est utilisé pour stocker les exécutables GATK, les scripts, les fichiers de carte d'alignement binaire (BAM), les fichiers de référence, les fichiers d'index, les fichiers de dictionnaire et les fichiers de sortie pour les appels de variantes.
Configuration et exécution de la GATK
Installez les prérequis suivants sur la VM RedHat Enterprise 8.3 Linux :
-
Java 8 ou SDK 1.8 ou version ultérieure
-
Télécharger GATK 4.2.0.0 depuis le large Institute "Site GitHub". Les données de séquence du génome sont généralement stockées sous la forme d'une série de colonnes ASCII délimitées par des tabulations. Cependant, l'espace ASCII est trop important pour être stocké. Par conséquent, un nouveau standard évolué appelé fichier BAM (*.bam). Un fichier BAM stocke les données de séquence sous forme compressée, indexée et binaire. Nous "téléchargé" Un ensemble de fichiers BAM disponibles publiquement pour l'exécution GATK à partir de l' "domaine public". Nous avons également téléchargé des fichiers d'index (\*.bai), des fichiers de dictionnaire (*. dict) et les fichiers de données de référence (*. fasta) du même domaine public.
Après le téléchargement, le kit d'outils GATK dispose d'un fichier jar et d'un ensemble de scripts de support.
-
gatk-package-4.2.0.0-local.jarexécutable -
gatkfichier de script.
Nous avons téléchargé les fichiers BAM et les fichiers d'index, de dictionnaire et de génome de référence correspondants pour une famille composée de fichiers de père, mère et fils *.bam.
Moteur Cromwell
Cromwell est un moteur open-source axé sur les flux de travail scientifiques qui permet la gestion des flux de travail. Le moteur Cromwell peut fonctionner en deux "modes", Mode serveur ou mode d'exécution d'un seul flux de travail. Le comportement du moteur Cromwell peut être contrôlé à l'aide du "Fichier de configuration du moteur Cromwell".
Nous utilisons le moteur Cromwell pour exécuter les flux de travail et les pipelines à grande échelle. Le moteur Cromwell est convivial "langage de description de workflow" Langage de script basé sur WDL. Cromwell prend également en charge une deuxième norme de script de flux de travail appelée langage de flux de travail commun (CWL). Tout au long de ce rapport technique, nous avons utilisé WDL. À l'origine, le WDL a été développé par le large institut pour les pipelines d'analyse du génome. L'utilisation des workflows WDL peut être mise en œuvre à l'aide de plusieurs stratégies, notamment :
-
Chaînage linéaire. comme le nom l'indique, la sortie de la tâche #1 est envoyée à la tâche #2 comme entrée.
-
Multi-in/out. Ceci est similaire à la chaînage linéaire dans le fait que chaque tâche peut avoir plusieurs sorties envoyées en entrée aux tâches suivantes.
-
Scatter-rassembler. il s'agit de l'une des stratégies d'intégration des applications d'entreprise les plus puissantes disponibles, surtout lorsqu'elle est utilisée dans une architecture basée sur des événements. Chaque tâche s'exécute de façon dissociée, et le résultat de chaque tâche est consolidé dans le résultat final.
Il existe trois étapes lorsque WDL est utilisé pour faire fonctionner GATK en mode autonome :
-
Valider la syntaxe à l'aide de
womtool.jar.[root@genomics1 ~]# java -jar womtool.jar validate ghplo.wdl
-
Générer des entrées JSON.
[root@genomics1 ~]# java -jar womtool.jar inputs ghplo.wdl > ghplo.json
-
Exécutez le flux de travail à l'aide du moteur Cromwell et
Cromwell.jar.[root@genomics1 ~]# java -jar cromwell.jar run ghplo.wdl –-inputs ghplo.json
Le GATK peut être exécuté à l'aide de plusieurs méthodes; ce document explore trois de ces méthodes.
Exécution de GATK à l'aide du fichier jar
Examinons l’exécution d’un pipeline d’appel variante unique à l’aide de l’appelant variant en haplotype.
[root@genomics1 ~]# java -Dsamjdk.use_async_io_read_samtools=false \ -Dsamjdk.use_async_io_write_samtools=true \ -Dsamjdk.use_async_io_write_tribble=false \ -Dsamjdk.compression_level=2 \ -jar /mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar \ HaplotypeCaller \ --input /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ --output workshop_1906_2-germline_bams_father.validation.vcf \ --reference /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta
Dans cette méthode d'exécution, nous utilisons le fichier JAR d'exécution locale GATK, une seule commande Java pour appeler le fichier jar, et nous transmettons plusieurs paramètres à la commande.
-
Ce paramètre indique que nous invoons le
HaplotypeCallervariante du pipeline appelant. -
-- inputSpécifie le fichier BAM en entrée. -
--outputspécifie le fichier de sortie de variante dans le format d'appel de variante (*.vcf) ("réf"). -
Avec le
--referenceparamètre, nous sommes en passe de passer un génome de référence.
Une fois exécuté, les détails de sortie se trouvent dans la section "Sortie pour l'exécution de GATK à l'aide du fichier jar."
Exécution de GATK à l'aide du script ./gatk
La trousse à outils GATK peut être exécutée à l'aide de l' ./gatk script. Examinons la commande suivante :
[root@genomics1 execution]# ./gatk \ --java-options "-Xmx4G" \ HaplotypeCaller \ -I /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ -R /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta \ -O /mnt/genomics/GATK/TEST\ DATA/variants.vcf
Nous transmettons plusieurs paramètres à la commande.
-
Ce paramètre indique que nous invoons le
HaplotypeCallervariante du pipeline appelant. -
-ISpécifie le fichier BAM en entrée. -
-Ospécifie le fichier de sortie de variante dans le format d'appel de variante (*.vcf) ("réf"). -
Avec le
-Rparamètre, nous sommes en passe de passer un génome de référence.
Une fois exécuté, les détails de sortie se trouvent dans la section
Exécution de la GATK à l'aide du moteur Cromwell
Nous utilisons le moteur Cromwell pour gérer l'exécution GATK. Examinons la ligne de commande et ses paramètres.
[root@genomics1 genomics]# java -jar cromwell-65.jar \ run /mnt/genomics/GATK/seq/ghplo.wdl \ --inputs /mnt/genomics/GATK/seq/ghplo.json
Ici, nous invoquons la commande Java en passant le -jar paramètre pour indiquer que nous avons l'intention d'exécuter un fichier jar, par exemple, Cromwell-65.jar. Le paramètre suivant a réussi (run) Indique que le moteur Cromwell fonctionne en mode RUN, l'autre option possible est le mode serveur. Le paramètre suivant est *.wdl Que le mode Exécuter doit utiliser pour exécuter les pipelines. Le paramètre suivant est l'ensemble des paramètres d'entrée des flux de travail exécutés.
Voici le contenu du ghplo.wdl type de fichier :
[root@genomics1 seq]# cat ghplo.wdl
workflow helloHaplotypeCaller {
call haplotypeCaller
}
task haplotypeCaller {
File GATK
File RefFasta
File RefIndex
File RefDict
String sampleName
File inputBAM
File bamIndex
command {
java -jar ${GATK} \
HaplotypeCaller \
-R ${RefFasta} \
-I ${inputBAM} \
-O ${sampleName}.raw.indels.snps.vcf
}
output {
File rawVCF = "${sampleName}.raw.indels.snps.vcf"
}
}
[root@genomics1 seq]#
Voici le fichier JSON correspondant avec les entrées du moteur Cromwell.
[root@genomics1 seq]# cat ghplo.json
{
"helloHaplotypeCaller.haplotypeCaller.GATK": "/mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar",
"helloHaplotypeCaller.haplotypeCaller.RefFasta": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta",
"helloHaplotypeCaller.haplotypeCaller.RefIndex": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta.fai",
"helloHaplotypeCaller.haplotypeCaller.RefDict": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.dict",
"helloHaplotypeCaller.haplotypeCaller.sampleName": "fatherbam",
"helloHaplotypeCaller.haplotypeCaller.inputBAM": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bam",
"helloHaplotypeCaller.haplotypeCaller.bamIndex": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bai"
}
[root@genomics1 seq]#
Veuillez noter que Cromwell utilise une base de données in-memory pour l'exécution. Une fois exécuté, le journal de sortie est visible dans la section "Sortie pour l'exécution de la GATK à l'aide du moteur Cromwell."
Pour un ensemble complet d'étapes sur la façon d'exécuter GATK, voir "Documentation GATK".