

Genomica - Installazione ed esecuzione di GATK
 Suggerisci modifiche
Suggerisci modifiche
Secondo il National Human Genome Research Institute ( "NHGRI"), "la genomica è lo studio di tutti i geni di una persona (il genoma), comprese le interazioni di questi geni tra loro e con l'ambiente di una persona. "
In base a. "NHGRI"L'acido desossiribonucleico (DNA) è il composto chimico che contiene le istruzioni necessarie per sviluppare e dirigere le attività di quasi tutti gli organismi viventi. Le molecole di DNA sono costituite da due trefoli torcenti accoppiati, spesso indicati come a doppia elica". "Il set completo di DNA di un organismo è chiamato genoma".
Il sequenziamento è il processo di determinazione dell'ordine esatto delle basi in un filamento di DNA. Uno dei tipi più comuni di sequenziamento oggi utilizzato è chiamato sequenziamento per sintesi. Questa tecnica utilizza l'emissione di segnali fluorescenti per ordinare le basi. I ricercatori possono utilizzare il sequenziamento del DNA per cercare variazioni genetiche e qualsiasi mutazione che possa svolgere un ruolo nello sviluppo o nella progressione di una malattia mentre una persona è ancora nello stadio embrionale.
Dall'identificazione del campione alla variante, all'annotazione e alla previsione
Ad alto livello, la genomica può essere classificata nei seguenti passaggi. Questo non è un elenco completo:
-
Raccolta dei campioni.
-
"Sequenziamento del genoma" utilizzo di un sequencer per generare i dati raw.
-
Pre-elaborazione. Ad esempio, "deduplica" utilizzo di "Picard".
-
Analisi genomica.
-
Mappatura a un genoma di riferimento.
-
"Variante" L'identificazione e l'annotazione vengono in genere eseguite utilizzando GATK e strumenti simili.
-
-
Integrazione nel sistema di cartelle cliniche elettroniche (EHR).
-
"Stratificazione della popolazione" e identificazione della variazione genetica attraverso la posizione geografica e il background etnico.
-
"Modelli predittivi" utilizzando un significativo polimorfismo a singolo nucleotide.
La figura seguente mostra il processo che va dal campionamento all'identificazione della variante, all'annotazione e alla previsione.
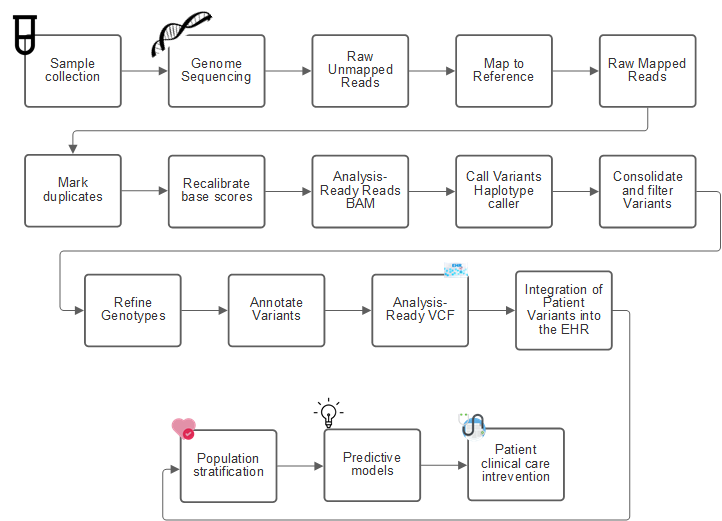
Il progetto Human Genome è stato completato nell'aprile 2003 e il progetto ha realizzato una simulazione di altissima qualità della sequenza di genomi umani disponibile in pubblico dominio. Questo genoma di riferimento ha dato inizio a un'esplosione nella ricerca e nello sviluppo delle capacità genomiche. Praticamente ogni disturbo umano ha una firma nei geni di quell'essere umano. Fino a poco tempo fa, i medici utilizzavano i geni per predire e determinare i difetti congeniti come l'anemia falciforme, causata da un certo schema di ereditarietà causato da un cambiamento in un singolo gene. Il tesoro dei dati messi a disposizione dal progetto sul genoma umano ha portato all'avvento dello stato attuale delle capacità genomiche.
La genomica offre una vasta gamma di vantaggi. Ecco una piccola serie di vantaggi nei settori sanitario e delle scienze biologiche:
-
Migliore diagnosi presso i punti di cura
-
Migliore prognosi
-
Medicina di precisione
-
Piani di trattamento personalizzati
-
Migliore monitoraggio delle malattie
-
Riduzione degli eventi avversi
-
Migliore accesso alle terapie
-
Miglioramento del monitoraggio delle malattie
-
Partecipazione efficace agli studi clinici e migliore selezione dei pazienti per gli studi clinici basati sui genotipi.
La genomica è un "bestia a quattro teste," a causa delle esigenze di calcolo per tutto il ciclo di vita di un set di dati: acquisizione, storage, distribuzione e analisi.
GATK (Genome Analysis Toolkit)
GATK è stata sviluppata come piattaforma per la data science presso "Broad Institute". GATK è un insieme di strumenti open-source che consentono l'analisi del genoma, in particolare rilevamento, identificazione, annotazione e genotipizzazione delle varianti. Uno dei vantaggi di GATK è che il set di strumenti e/o comandi può essere concatenato per formare un workflow completo. Le principali sfide affrontate da un ampio istituto sono le seguenti:
-
Comprendere le cause alla radice e i meccanismi biologici delle malattie.
-
Identificare gli interventi terapeutici che agiscono alla causa fondamentale di una malattia.
-
Comprendere la linea di vista dalle varianti al funzionamento in fisiologia umana.
-
Creare standard e policy "framework" per la rappresentazione dei dati genoma, lo storage, l'analisi, la sicurezza e così via.
-
Standardizzare e socializzare database di aggregazione dei genomi interoperabili (gnomAD).
-
Monitoraggio, diagnosi e trattamento dei pazienti basati sul genoma con maggiore precisione.
-
Aiuta a implementare strumenti che prevedano le malattie ben prima che appaiano i sintomi.
-
Crea e potenzia una community di collaboratori interdisciplinari per affrontare i problemi più difficili e importanti della biomedicina.
Secondo il GATK e l'ampio istituto, il sequenziamento del genoma deve essere trattato come un protocollo in un laboratorio di patologia; ogni attività è ben documentata, ottimizzata, riproducibile e coerente tra campioni ed esperimenti. Di seguito viene riportata una serie di procedure consigliate dal Broad Institute. Per ulteriori informazioni, vedere "Sito web di GATK".
Configurazione di FlexPod
La convalida del carico di lavoro di genomics include una configurazione da zero di una piattaforma di infrastruttura FlexPod. La piattaforma FlexPod è altamente disponibile e può essere scalata in modo indipendente; ad esempio, la rete, lo storage e il calcolo possono essere scalati in modo indipendente. Abbiamo utilizzato la seguente guida alla progettazione convalidata da Cisco come documento di riferimento sull'architettura per configurare l'ambiente FlexPod: "Data center FlexPod con VMware vSphere 7.0 e NetApp ONTAP 9.7". Scopri i seguenti punti salienti della configurazione della piattaforma FlexPod:
Per eseguire la configurazione del laboratorio FlexPod, attenersi alla seguente procedura:
-
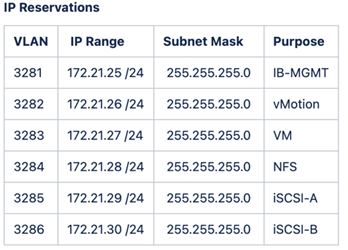
La configurazione e la convalida del laboratorio FlexPod utilizza le seguenti prenotazioni IP4 e VLAN.
-
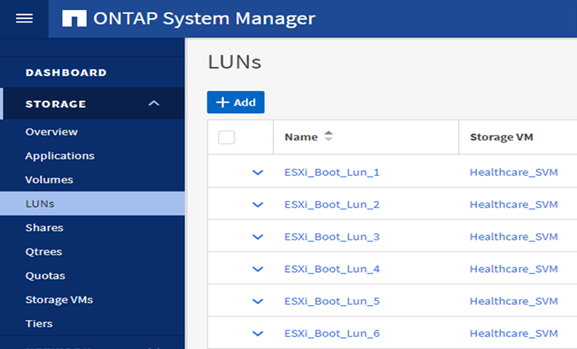
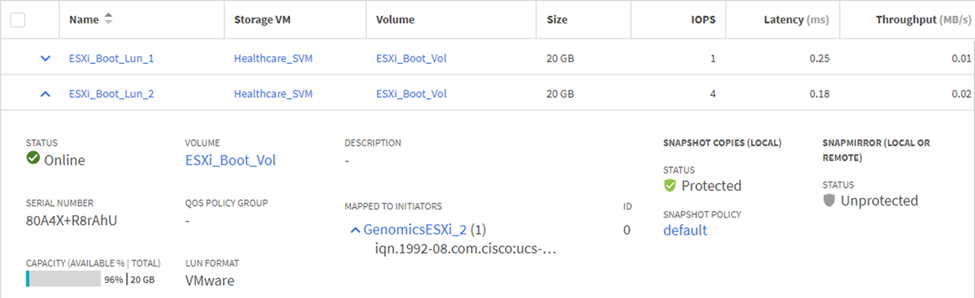
Configurare le LUN di avvio basate su iSCSI sulla SVM ONTAP.
-
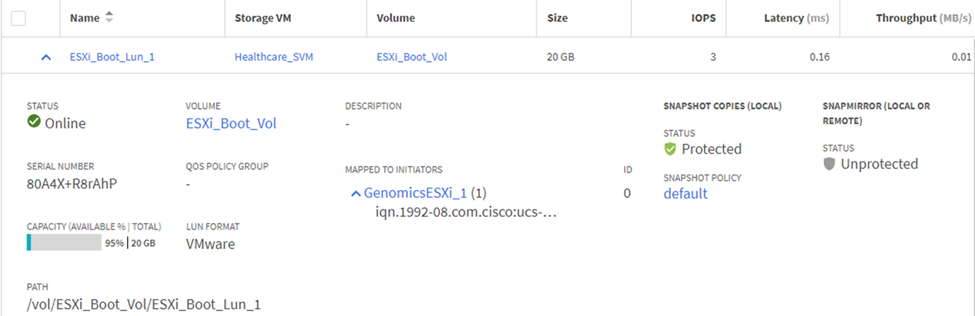
Mappare i LUN ai gruppi di iniziatori iSCSI.
-
Installare vSphere 7.0 con l'avvio iSCSI.
-
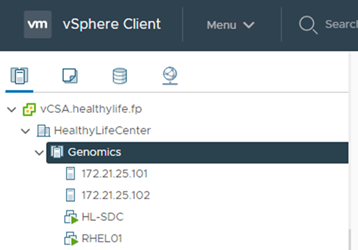
Registrare gli host ESXi con vCenter.
-
Eseguire il provisioning di un datastore NFS
infra_datastore_nfsSullo storage ONTAP.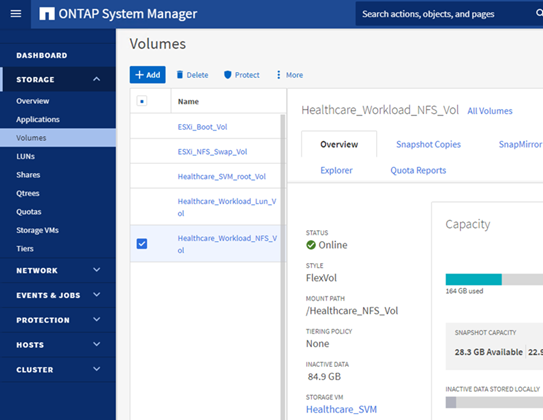
-
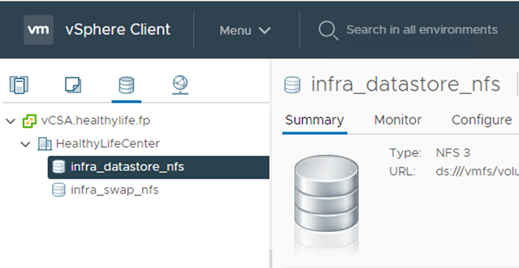
Aggiungere il datastore al vCenter.
-
Utilizzando vCenter, aggiungere un datastore NFS agli host ESXi.
-
Utilizzando vCenter, creare una macchina virtuale Red Hat Enterprise Linux (RHEL) 8.3 per eseguire GATK.
-
Un datastore NFS viene presentato alla macchina virtuale e montato su
/mnt/genomics, Utilizzato per memorizzare file eseguibili GATK, script, file BAM (Binary Alignment Map), file di riferimento, file di indice, file del dizionario e file out per la chiamata delle varianti.
Configurazione ed esecuzione di GATK
Installare i seguenti prerequisiti su RedHat Enterprise 8.3 Linux VM:
-
Java 8 o SDK 1.8 o versione successiva
-
Scarica GATK 4.2.0.0 dal Broad Institute "Sito GitHub". I dati della sequenza genoma sono generalmente memorizzati sotto forma di una serie di colonne ASCII delimitate da tabulazioni. Tuttavia, ASCII occupa troppo spazio per la memorizzazione. Pertanto, un nuovo standard evoluto chiamato file BAM (.bam). Un file BAM memorizza i dati della sequenza in un formato compresso, indicizzato e binario. Noi "scaricato" Un insieme di file BAM disponibili pubblicamente per l'esecuzione di GATK da "di dominio pubblico". Abbiamo anche scaricato file di indice (.bai), file di dizionario (. dict) e file di dati di riferimento (. fasta) dello stesso dominio pubblico.
Dopo il download, il kit di strumenti GATK ha un file jar e una serie di script di supporto.
-
gatk-package-4.2.0.0-local.jareseguibile -
gatkfile di script.
Abbiamo scaricato i file BAM e i corrispondenti file di indice, dizionario e genoma di riferimento per una famiglia composta da file *.bam padre, madre e figlio.
Motore Cromwell
Cromwell è un motore open-source orientato ai flussi di lavoro scientifici che consente la gestione del workflow. Il motore Cromwell può essere eseguito in due "modalità", Server mode o Run mode a singolo flusso di lavoro. Il comportamento del motore Cromwell può essere controllato tramite "File di configurazione del motore Cromwell".
-
Server mode. attiva "Riposante" Esecuzione dei flussi di lavoro nel motore Cromwell.
-
Run mode. la modalità Run è più adatta per l'esecuzione di singoli flussi di lavoro in Cromwell, "rif" Per una serie completa di opzioni disponibili in modalità Run.
Utilizziamo il motore Cromwell per eseguire flussi di lavoro e pipeline su larga scala. Il motore Cromwell utilizza un sistema intuitivo "linguaggio di descrizione del workflow" Linguaggio di scripting basato su (WDL). Cromwell supporta anche un secondo standard di scripting per il workflow, denominato Common workflow Language (CWL). Nel corso di questo report tecnico, abbiamo utilizzato WDL. WDL è stato originariamente sviluppato dal Broad Institute for Genome analysis Pipeline. I flussi di lavoro WDL possono essere implementati utilizzando diverse strategie, tra cui:
-
Linear Chaining. come suggerisce il nome, l'output dell'attività n. 1 viene inviato all'attività n. 2 come input.
-
Multi-in/out. questo è simile al concatenamento lineare in quanto ogni task può avere più output inviati come input a task successivi.
-
Scatter-Gather. si tratta di una delle strategie di integrazione applicativa aziendale (EAI) più potenti disponibili, soprattutto se utilizzata in un'architettura basata sugli eventi. Ogni task viene eseguito in modo disaccoppiato e l'output di ogni task viene consolidato nell'output finale.
Quando si utilizza WDL per eseguire GATK in una modalità standalone, sono disponibili tre passaggi:
-
Validare la sintassi utilizzando
womtool.jar.[root@genomics1 ~]# java -jar womtool.jar validate ghplo.wdl
-
Generare input JSON.
[root@genomics1 ~]# java -jar womtool.jar inputs ghplo.wdl > ghplo.json
-
Eseguire il flusso di lavoro utilizzando il motore Cromwell e.
Cromwell.jar.[root@genomics1 ~]# java -jar cromwell.jar run ghplo.wdl –-inputs ghplo.json
Il GATK può essere eseguito utilizzando diversi metodi; questo documento esplora tre di questi metodi.
Esecuzione di GATK utilizzando il file jar
Esaminiamo ora l'esecuzione di una singola pipeline di chiamate con il chiamante della variante haplotype.
[root@genomics1 ~]# java -Dsamjdk.use_async_io_read_samtools=false \ -Dsamjdk.use_async_io_write_samtools=true \ -Dsamjdk.use_async_io_write_tribble=false \ -Dsamjdk.compression_level=2 \ -jar /mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar \ HaplotypeCaller \ --input /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ --output workshop_1906_2-germline_bams_father.validation.vcf \ --reference /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta
In questo metodo di esecuzione, utilizziamo il file jar di esecuzione locale di GATK, utilizziamo un singolo comando java per richiamare il file jar e passiamo diversi parametri al comando.
-
Questo parametro indica che stiamo richiamando
HaplotypeCallerpipeline chiamante variante. -
-- inputSpecifica il file BAM di input. -
--outputspecifica il file di output della variante nel formato di chiamata della variante (*.vcf) ("rif"). -
Con
--referenceparametro, stiamo passando un genoma di riferimento.
Una volta eseguita l'operazione, i dettagli dell'output sono disponibili nella sezione "Output per l'esecuzione di GATK utilizzando il file jar."
Esecuzione di GATK utilizzando lo script ./gatk
Il kit di strumenti GATK può essere eseguito utilizzando ./gatk script. Esaminiamo il seguente comando:
[root@genomics1 execution]# ./gatk \ --java-options "-Xmx4G" \ HaplotypeCaller \ -I /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ -R /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta \ -O /mnt/genomics/GATK/TEST\ DATA/variants.vcf
Passiamo diversi parametri al comando.
-
Questo parametro indica che stiamo richiamando
HaplotypeCallerpipeline chiamante variante. -
-ISpecifica il file BAM di input. -
-Ospecifica il file di output della variante nel formato di chiamata della variante (*.vcf) ("rif"). -
Con
-Rparametro, stiamo passando un genoma di riferimento.
Una volta eseguita l'operazione, i dettagli dell'output sono disponibili nella sezione
Esecuzione di GATK utilizzando il motore Cromwell
Utilizziamo il motore Cromwell per gestire l'esecuzione di GATK. Esaminiamo la riga di comando e i relativi parametri.
[root@genomics1 genomics]# java -jar cromwell-65.jar \ run /mnt/genomics/GATK/seq/ghplo.wdl \ --inputs /mnt/genomics/GATK/seq/ghplo.json
In questo caso, viene richiamato il comando Java passando a. -jar parametro per indicare che si intende eseguire un file jar, ad esempio Cromwell-65.jar. Il parametro successivo è stato superato (run) Indica che il motore Cromwell è in esecuzione in modalità Run, mentre l'altra opzione possibile è la modalità Server. Il parametro successivo è *.wdl Che la modalità Run debba utilizzare per eseguire le pipeline. Il parametro successivo è l'insieme di parametri di input per i flussi di lavoro in esecuzione.
Di seguito sono elencati i contenuti di ghplo.wdl file simile a:
[root@genomics1 seq]# cat ghplo.wdl
workflow helloHaplotypeCaller {
call haplotypeCaller
}
task haplotypeCaller {
File GATK
File RefFasta
File RefIndex
File RefDict
String sampleName
File inputBAM
File bamIndex
command {
java -jar ${GATK} \
HaplotypeCaller \
-R ${RefFasta} \
-I ${inputBAM} \
-O ${sampleName}.raw.indels.snps.vcf
}
output {
File rawVCF = "${sampleName}.raw.indels.snps.vcf"
}
}
[root@genomics1 seq]#
Ecco il file JSON corrispondente con gli input al motore Cromwell.
[root@genomics1 seq]# cat ghplo.json
{
"helloHaplotypeCaller.haplotypeCaller.GATK": "/mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar",
"helloHaplotypeCaller.haplotypeCaller.RefFasta": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta",
"helloHaplotypeCaller.haplotypeCaller.RefIndex": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta.fai",
"helloHaplotypeCaller.haplotypeCaller.RefDict": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.dict",
"helloHaplotypeCaller.haplotypeCaller.sampleName": "fatherbam",
"helloHaplotypeCaller.haplotypeCaller.inputBAM": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bam",
"helloHaplotypeCaller.haplotypeCaller.bamIndex": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bai"
}
[root@genomics1 seq]#
Tenere presente che Cromwell utilizza un database in-memory per l'esecuzione. Una volta eseguito, il log di output viene visualizzato nella sezione "Output per l'esecuzione di GATK utilizzando il motore Cromwell."
Per una serie completa di passaggi su come eseguire GATK, vedere "Documentazione GATK".