

ゲノム - GATK のセットアップと実行
 変更を提案
変更を提案
国立ヒトゲノム研究所によると "NHGRI") 、「ゲノムとは、人の遺伝子 ( ゲノム ) のすべての研究であり、これらの遺伝子相互作用や人の環境との相互作用を含みます。」
に従って "NHGRI"「デオキシリボヌクリク酸( DNA )は、ほぼすべての生物の活動を開発し、誘導するために必要な指示を含む化学化合物です。DNA 分子は、二重らせんと呼ばれる 2 つのツイスト、ペアストランドで構成されています。」 「生物の DNA の完全なセットは、ゲノムと呼ばれています。」
配列決定は DNA の鎖の塩基の正確な順序を決定するプロセスである。現在使用されている最も一般的なシーケンスタイプの 1 つは、合成による順序付けと呼ばれます。この技術では、蛍光信号の放射を使用して塩基を並べます。研究者は DNA シーケンシングを使用して、遺伝子変異や、人がまだ初期段階にある間に疾患の発症または進行に関与する可能性のある突然変異を検索することができる。
サンプルからバリアントの識別、注釈、および予測まで
ゲノム解析の概要は、以下のステップに分類できます。これは完全なリストではありません。
次の図に、サンプリングからバリアントの識別、注釈、および予測までのプロセスを示します。
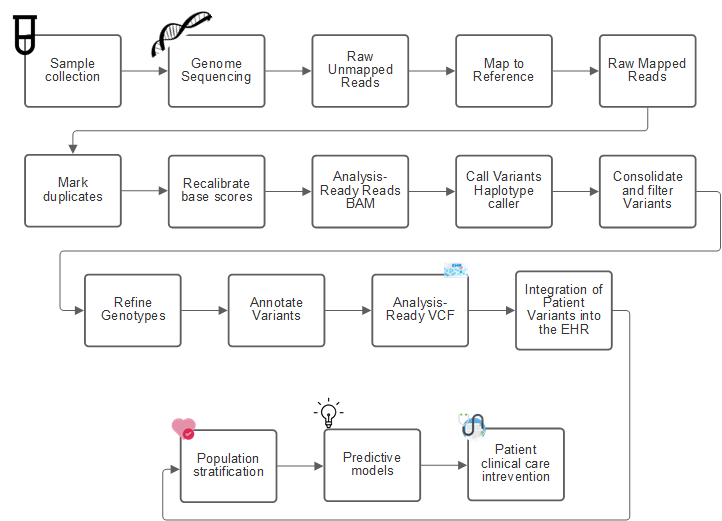
ヒトゲノム計画は 2003 年 4 月に完成し、このプロジェクトは、パブリックドメインで利用可能なヒトゲノム配列を非常に高品質でシミュレーションしました。このリファレンスゲノムは、ゲノム機能の研究開発で爆発的に増加しました。事実上すべての人間の病気にその人間の遺伝子の署名がある。最近まで、医師は、単一遺伝子の変化によって発生した特定の相続パターンによって引き起こされる、鎌状赤血球貧血などの出生異常を予測し、判定するために遺伝子を利用していた。ヒトゲノムプロジェクトで収集された膨大なデータがゲノム機能の最新状態に登場しました。
ゲノミクスには幅広いメリットがあります。ヘルスケアおよびライフサイエンス分野のメリットを以下に示します。
-
治療時点でのより良い診断
-
予後が良好である
-
精密医学
-
パーソナライズされた治療計画
-
疾患モニタリングの向上
-
有害事象の減少
-
治療へのアクセスが向上しました
-
疾患モニタリングの改善
-
有効な臨床試験への参加と、遺伝子型に基づく臨床試験の患者の選択の向上。
ゲノミクスは a "4 つのヘッドを持つ獣、" 取得、ストレージ、分散、分析という、データセットのライフサイクル全体にわたるコンピューティングのニーズがあるためです。
ゲノム解析ツールキット( GATK )
GATK は、でデータサイエンスプラットフォームとして開発されました "ブロードインスティテュート"。GATK は、ゲノム解析を可能にする一連のオープンソース・ツールで、特に変異検出、同定、アノテーション、ジェノタイピングなどを行います。GATK の利点の 1 つは、ツールやコマンドのセットを連鎖させて、完全なワークフローを形成できることです。ブロード研究所が取り組む主な課題は、次のとおりです。
-
病気の根本原因と生物学的メカニズムを理解する。
-
疾患の基礎原因で作用する治療的介入を特定する。
-
変異体から人間の生理学的な機能まで、視線を理解します。
-
標準とポリシーを作成します "フレームワーク" ゲノムデータの表示、保存、分析、セキュリティなどを行います。
-
相互運用可能なゲノム集約データベース( gnomad )を標準化し、社会化します。
-
ゲノムを用いたモニタリング、診断、および患者の治療をより正確に行うことができます。
-
症状が現れる前に疾患を適切に予測するツールの導入を支援します。
-
生物医学における最も困難で最も重要な問題に対処するために、学際的な協力者のコミュニティを作成し、強化します。
GATK と The Broad Institute によると、ゲノム配列決定は病理学ラボでプロトコルとして扱われるべきです。どのような作業でも、サンプルや実験全体でよく文書化され、最適化され、再現性があり、一貫性が保たれます。以下は、ブロード研究所が推奨する一連の手順です。詳細については、を参照してください "GATK の Web サイト"。
FlexPod セットアップ
ゲノミクスワークロードの検証には、 FlexPod インフラプラットフォームのスクラッチからのセットアップが含まれています。FlexPod プラットフォームは高可用性を備えており、個別に拡張できます。たとえば、ネットワーク、ストレージ、コンピューティングを個別に拡張できます。FlexPod 環境をセットアップするためのリファレンスアーキテクチャドキュメントとして、次のシスコ検証済み設計ガイドを使用しました。 "FlexPod Datacenter with VMware vSphere 7.0 and NetApp ONTAP 9.7 』を参照してください"。次の FlexPod プラットフォームのセットアップのハイライトを参照してください。
FlexPod のラボセットアップを実行するには、次の手順を実行します。
-
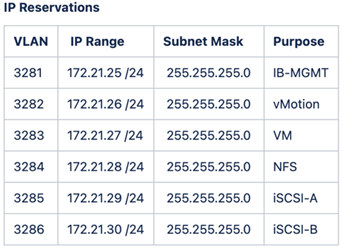
FlexPod ラボのセットアップと検証では、次の IP4 予約と VLAN を使用します。
-
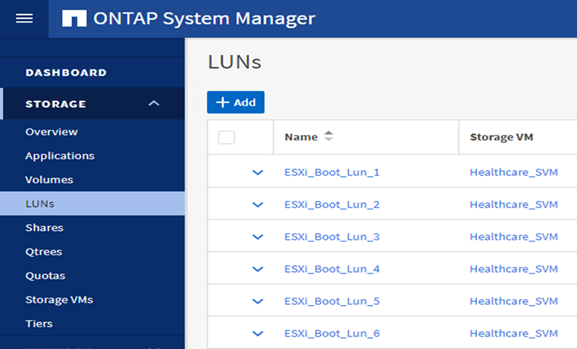
ONTAP SVM で iSCSI ベースのブート LUN を設定
-
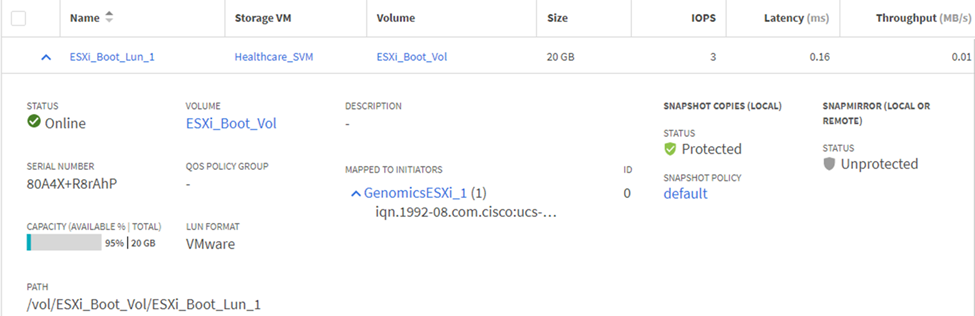
LUN を iSCSI イニシエータグループにマッピングします。
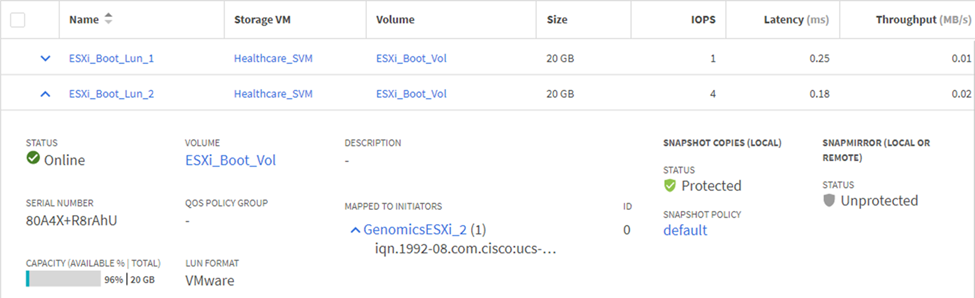
-
iSCSI ブートを使用して vSphere 7.0 をインストールします。
-
ESXi ホストを vCenter に登録します。
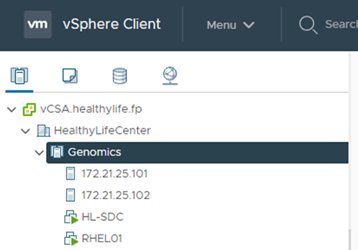
-
ONTAP ストレージ上で NFS データストア「 infra_datastore_nfs 」をプロビジョニングします。
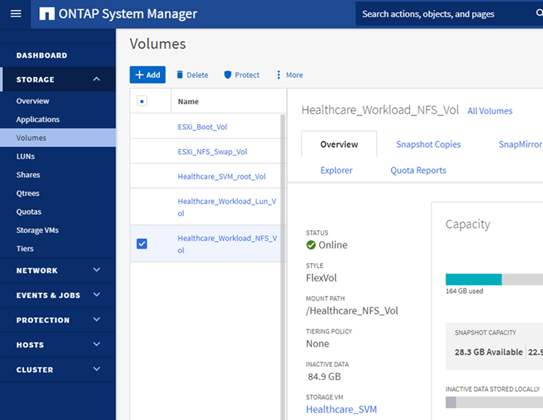
-
vCenter にデータストアを追加します。
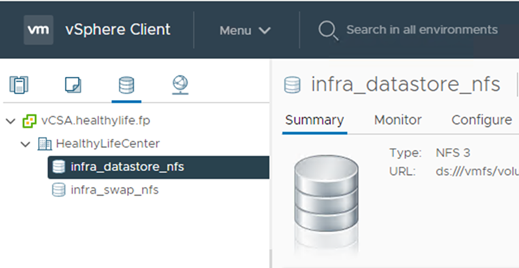
-
vCenter を使用して、 ESXi ホストに NFS データストアを追加します。
-
vCenter を使用して、 GATK を実行する Red Hat Enterprise Linux ( RHEL ) 8.3 VM を作成します。
-
NFS データストアが VM に提供され、「 /mnt/ゲノミクス 」でマウントされます。これは、 GATK 実行可能ファイル、スクリプト、 BAM (バイナリ・アライメント・マップ)ファイル、参照ファイル、インデックスファイル、辞書ファイル、およびバリアント呼び出し用の出力ファイルを格納するために使用されます。

GATK のセットアップと実行
Red Hat Enterprise 8.3 Linux VM に次の前提条件をインストールします。
-
Java 8 または SDK 1.8 以降
-
Broad Institute から GATK 4.2.0.0 をダウンロードしてください "GitHub サイト"。一般に、ゲノム配列データは、タブ区切りの一連の ASCII カラムの形で保存されます。ただし、 ASCII の保存に必要なスペースが多すぎます。したがって、新しい標準は BAM (*.bAM) ファイルと呼ばれて進化しました。BAM ファイルは、シーケンスデータを圧縮、インデックス化、およびバイナリ形式で格納します。私たち "ダウンロードしました" から GATK を実行するために公開されている BAM ファイルのセット "パブリックドメイン"。インデックスファイル( \*.bai )、辞書ファイル( \* )もダウンロードしました。dict )、および参照データファイル( * 。FASTA )を参照してください。
ダウンロード後、 GATK ツールキットには jar ファイルと一連のサポートスクリプトがあります。
-
GATK-PACKPACK-4.2.0.0 -local.jar 実行可能ファイル
-
「 GATK 」スクリプトファイル。
父、母、息子 * 。 BAM ファイルで構成された家族の BAM ファイルと対応する索引、辞書、参照ゲノムファイルをダウンロードしました。
クロムウェルエンジン
Cromwell は、ワークフロー管理を可能にする科学的なワークフローを対象としたオープンソースエンジンです。クロムウェルエンジンは 2 つの方法で作動できます "モード"、サーバーモード、または単一ワークフローの実行モード。クロムウェルエンジンの動作は、を使用して制御できます "クロムウェルエンジンコンフィギュレーションファイル"。
-
* サーバーモード。 * 有効にします "RESTful なホテル" クロムウェルエンジンでのワークフローの実行。
-
* 実行モード。 * 実行モードはクロムウェルで単一のワークフローを実行する場合に最適です。 "参照( Ref )" 実行モードで使用可能なすべてのオプションを表示します。
当社では、 Cromwell エンジンを使用してワークフローとパイプラインを大規模に実行しています。クロムウェルエンジンは使いやすいエンジンです "Workflow 概要の言語" (WDL) ベースのスクリプト言語。また、 Cromwell は、 Common Workflow Language ( CWL )と呼ばれる 2 つ目のワークフロースクリプト標準もサポートしています。このテクニカルレポートでは、 WDL を使用しました。WDL は、もともと、広範なゲノム解析パイプライン研究所によって開発されたものです。WDL ワークフローを使用するには、次のようないくつかの戦略を使用します。
-
* リニアチェーン。 * 名前が示すように、タスク #1 からの出力がタスク #2 に入力として送信されます。
-
* マルチイン / アウト。 * これは、各タスクで複数の出力を後続のタスクに入力として送信できる点で、リニアチェーンと似ています。
-
* Scatter-Gather * これは、特にイベント駆動型アーキテクチャで使用される場合に、最も強力なエンタープライズ・アプリケーション・インテグレーション( EAI )戦略の 1 つです。各タスクは分離された方法で実行され、各タスクの出力が最終出力に統合されます。
WDL を使用してスタンドアロンモードで GATK を実行するには、次の 3 つの手順があります。
-
「 womtool.jar 」を使用して構文を検証します。
[root@genomics1 ~]# java -jar womtool.jar validate ghplo.wdl
-
JSON の生成
[root@genomics1 ~]# java -jar womtool.jar inputs ghplo.wdl > ghplo.json
-
Cromwell エンジンと Cromwell.jar を使用してワークフローを実行します
[root@genomics1 ~]# java -jar cromwell.jar run ghplo.wdl –-inputs ghplo.json
GATK は、いくつかのメソッドを使用して実行できます。このドキュメントでは、これらの方法のうちの 3 つについて説明します。
jar ファイルを使用した GATK の実行
では、 hplotype バリアントの呼び出し側を使用した単一バリアントのコールパイプラインの実行について見てみましょう。
[root@genomics1 ~]# java -Dsamjdk.use_async_io_read_samtools=false \ -Dsamjdk.use_async_io_write_samtools=true \ -Dsamjdk.use_async_io_write_tribble=false \ -Dsamjdk.compression_level=2 \ -jar /mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar \ HaplotypeCaller \ --input /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ --output workshop_1906_2-germline_bams_father.validation.vcf \ --reference /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta
この実行方法では、 GATK ローカル実行 jar ファイルを使用し、 1 つの Java コマンドを使用して jar ファイルを呼び出します。このコマンドには、いくつかのパラメータが渡されます。
-
このパラメータは 'HaplotypeCaller バリアントの呼び出し側パイプラインを呼び出していることを示します
-
--input' は、入力 BAM ファイルを指定します。
-
--output' は、 variant 呼び出し形式( *.VCF )でバリアント出力ファイルを指定します。 ("参照( Ref )")。
-
「 --reference 」パラメータを使用して、参照ゲノムを渡しています。
実行すると、出力の詳細がセクションに表示されます "jar ファイルを使用して GATK を実行するための出力。"
./GATK スクリプトを使用した GATK の実行
GATK ツール・キットは './GATK' スクリプトを使用して実行できます次のコマンドを見てみましょう。
[root@genomics1 execution]# ./gatk \ --java-options "-Xmx4G" \ HaplotypeCaller \ -I /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ -R /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta \ -O /mnt/genomics/GATK/TEST\ DATA/variants.vcf
コマンドにはいくつかのパラメータを渡します。
-
このパラメータは 'HaplotypeCaller バリアントの呼び出し側パイプラインを呼び出していることを示します
-
「 -i 」は、入力 BAM ファイルを指定します。
-
「 -O 」は、バリアント・コール・フォーマット( *.VCF )でバリアント出力ファイルを指定します。 ("参照( Ref )")。
-
R パラメータを使用して、参照ゲノムを渡しています。
実行すると、出力の詳細がセクションに表示されます "016e203cf9beada735f224ab14d0b3af"
クロムウェルエンジンを使用した GATK の実行
当社では、クルムウェルエンジンを使用して GATK の実行を管理しています。コマンドラインとパラメータを見てみましょう。
[root@genomics1 genomics]# java -jar cromwell-65.jar \ run /mnt/genomics/GATK/seq/ghplo.wdl \ --inputs /mnt/genomics/GATK/seq/ghplo.json
ここでは '-car' パラメータを渡して java コマンドを呼び出しますこれは 'Cromwell-65.jar などの jar ファイルを実行することを示します次に渡されるパラメータ (`run') は、クロムウェルエンジンが実行モードで実行されていることを示します。もう 1 つのオプションはサーバーモードです。次のパラメータは '*.wdl ですこれは ' 実行モードがパイプラインを実行するために使用する必要があります次のパラメータは、実行するワークフローへの入力パラメータのセットです。
「 ghplo.wdl 」ファイルの内容は次のようになります。
[root@genomics1 seq]# cat ghplo.wdl
workflow helloHaplotypeCaller {
call haplotypeCaller
}
task haplotypeCaller {
File GATK
File RefFasta
File RefIndex
File RefDict
String sampleName
File inputBAM
File bamIndex
command {
java -jar ${GATK} \
HaplotypeCaller \
-R ${RefFasta} \
-I ${inputBAM} \
-O ${sampleName}.raw.indels.snps.vcf
}
output {
File rawVCF = "${sampleName}.raw.indels.snps.vcf"
}
}
[root@genomics1 seq]#
ここでは、 Cromwell エンジンへの入力を持つ、対応する JSON ファイルを示します。
[root@genomics1 seq]# cat ghplo.json
{
"helloHaplotypeCaller.haplotypeCaller.GATK": "/mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar",
"helloHaplotypeCaller.haplotypeCaller.RefFasta": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta",
"helloHaplotypeCaller.haplotypeCaller.RefIndex": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta.fai",
"helloHaplotypeCaller.haplotypeCaller.RefDict": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.dict",
"helloHaplotypeCaller.haplotypeCaller.sampleName": "fatherbam",
"helloHaplotypeCaller.haplotypeCaller.inputBAM": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bam",
"helloHaplotypeCaller.haplotypeCaller.bamIndex": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bai"
}
[root@genomics1 seq]#
Cromwell は実行にインメモリデータベースを使用していることに注意してください。実行すると、出力ログがセクションに表示されます "クロムウェルエンジンを使用した GATK 実行用出力。"
GATK を実行するための包括的な手順については、を参照してください "GATK のドキュメント"。