

Genômica - configuração e execução do GATK
 Sugerir alterações
Sugerir alterações
De acordo com o National Human Genome Research Institute ( "NHGRI"), "a genômica é o estudo de todos os genes de uma pessoa (o genoma), incluindo interações desses genes entre si e com o ambiente de uma pessoa. "
De acordo com o "NHGRI", "o ácido desoxirribonucleico (DNA) é o composto químico que contém as instruções necessárias para desenvolver e direcionar as atividades de quase todos os organismos vivos. As moléculas de DNA são feitas de dois fios torcidos, emparelhados, muitas vezes referidos como uma hélice dupla." "O conjunto completo de DNA de um organismo é chamado de genoma."
Sequenciamento é o processo de determinação da ordem exata das bases em uma cadeia de DNA. Um dos tipos mais comuns de sequenciamento usado hoje é chamado sequenciamento por síntese. Esta técnica utiliza a emissão de sinais fluorescentes para ordenar as bases. Os pesquisadores podem usar o sequenciamento de DNA para procurar variações genéticas e quaisquer mutações que possam desempenhar um papel no desenvolvimento ou progressão de uma doença enquanto uma pessoa ainda está em estágio embrionário.
Da amostra à identificação, anotação e previsão da variante
Em um nível elevado, a genômica pode ser classificada nas seguintes etapas. Esta não é uma lista exaustiva:
-
Colheita de amostras.
-
"Sequenciamento do genoma" usando um sequenciador para gerar os dados brutos.
-
Pré-processamento. Por exemplo "deduplicação", usando "Picard".
-
Análise genômica.
-
Mapeamento para um genoma de referência.
-
"Variante" Identificação e anotação normalmente realizadas usando o GATK e ferramentas semelhantes.
-
-
Integração no sistema de registo eletrónico de saúde (EHR).
-
"Estratificação populacional" e identificação da variação genética entre localização geográfica e origem étnica.
-
"Modelos preditivos" usando polimorfismo de um único nucleotídeo significativo.
A figura a seguir mostra o processo de amostragem à identificação, anotação e previsão da variante.
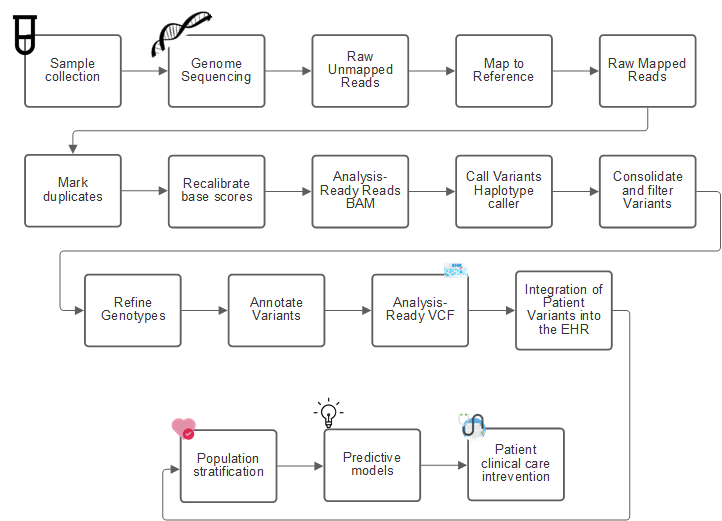
O projeto Genoma humano foi concluído em abril de 2003, e o projeto fez uma simulação de alta qualidade da sequência do genoma humano disponível no domínio público. Este genoma de referência iniciou uma explosão na pesquisa e desenvolvimento de capacidades genômicas. Praticamente toda doença humana tem uma assinatura nos genes desse ser humano. Até recentemente, os médicos estavam alavancando genes para prever e determinar defeitos congênitos como anemia falciforme, que é causada por um certo padrão de herança causado por uma mudança em um único gene. O tesouro dos dados que estão sendo disponibilizados pelo projeto do genoma humano levou ao advento do estado atual das capacidades genômicas.
A genômica tem um amplo conjunto de benefícios. Aqui está um pequeno conjunto de benefícios nos domínios da saúde e ciências da vida:
-
Melhor diagnóstico no ponto de atendimento
-
Melhor prognóstico
-
Medicina de precisão
-
Planos de tratamento personalizados
-
Melhor monitoramento de doenças
-
Redução de eventos adversos
-
Melhor acesso a terapias
-
Melhor monitoramento de doenças
-
Participação efetiva em ensaios clínicos e melhor seleção de pacientes para ensaios clínicos com base em genótipos.
A genômica ocorre "besta de quatro cabeças," devido às demandas computacionais em todo o ciclo de vida de um conjunto de dados: Aquisição, storage, distribuição e análise.
Genoma Analysis Toolkit (GATK)
O GATK foi desenvolvido como uma plataforma de ciência de dados na "Instituto amplo". O GATK é um conjunto de ferramentas de código aberto que permitem a análise do genoma, especificamente descoberta de variantes, identificação, anotação e genotipagem. Um dos benefícios do GATK é que o conjunto de ferramentas e comandos ou podem ser encadeados para formar um fluxo de trabalho completo. Os principais desafios que o instituto amplo enfrenta são os seguintes:
-
Compreender as causas e os mecanismos biológicos das doenças.
-
Identificar intervenções terapêuticas que atuam na causa fundamental de uma doença.
-
Entenda a linha de visão de variantes para funcionar na fisiologia humana.
-
Crie padrões e políticas "frameworks" para representação de dados do genoma, armazenamento, análise, segurança e assim por diante.
-
Padronize e socialize bancos de dados interoperáveis de agregação de genoma (gnomAD).
-
Monitoramento, diagnóstico e tratamento baseados em genoma de pacientes com maior precisão.
-
Ajude a implementar ferramentas que predizem doenças bem antes que os sintomas apareçam.
-
Criar e capacitar uma comunidade de colaboradores multidisciplinares para ajudar a resolver os problemas mais difíceis e mais importantes da biomedicina.
De acordo com o GATK e o instituto amplo, o sequenciamento do genoma deve ser Tratado como um protocolo em um laboratório de patologia; cada tarefa é bem documentada, otimizada, reprodutível e consistente em amostras e experimentos. A seguir está um conjunto de etapas recomendadas pelo Broad Institute. Para obter mais informações, consulte o "Website da GATK".
Configuração FlexPod
A validação da carga de trabalho genômica inclui uma configuração do zero de uma plataforma de infraestrutura FlexPod. A plataforma FlexPod é altamente disponível e pode ser dimensionada de forma independente. Por exemplo, a rede, o storage e a computação podem ser dimensionados de forma independente. Usamos o seguinte guia de design validado pela Cisco como o documento de arquitetura de referência para configurar o ambiente FlexPod: "Data center FlexPod com VMware vSphere 7,0 e NetApp ONTAP 9 .7". Veja os seguintes destaques de configuração da plataforma FlexPod:
Para executar a configuração do laboratório do FlexPod, execute as seguintes etapas:
-
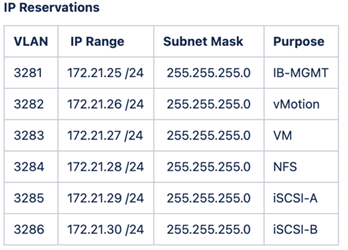
A configuração e a validação do laboratório FlexPod usam as seguintes reservas e VLANs IP4.1X.
-
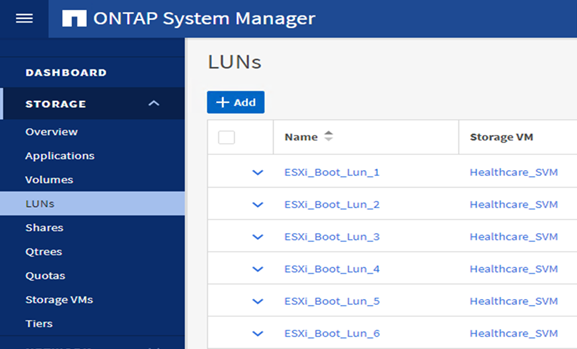
Configurar LUNs de inicialização baseados em iSCSI no ONTAP SVM.
-
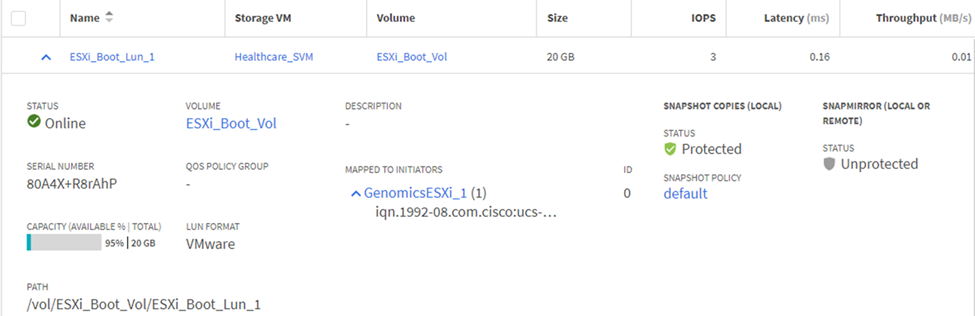
Mapear LUNs para grupos de iniciadores iSCSI.
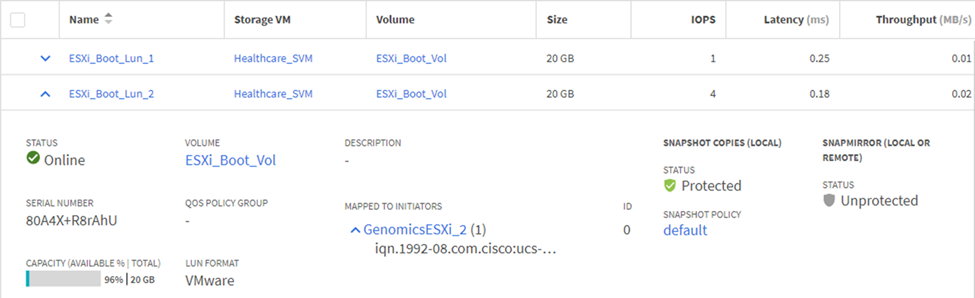
-
Instale o vSphere 7,0 com a inicialização iSCSI.
-
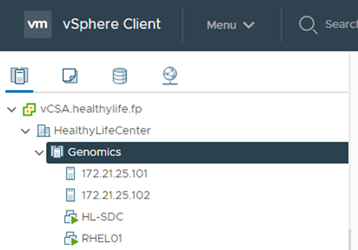
Registre hosts ESXi com o vCenter.
-
Provisionar um armazenamento de dados NFS
infra_datastore_nfsno storage ONTAP.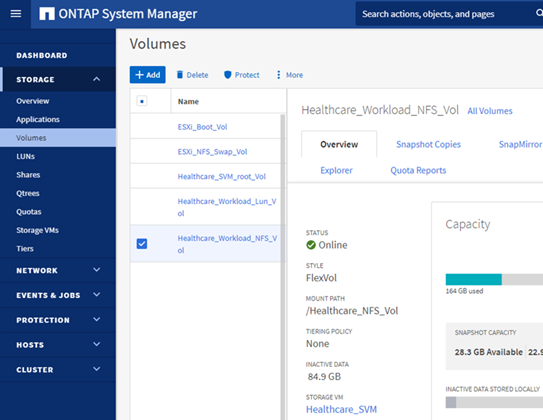
-
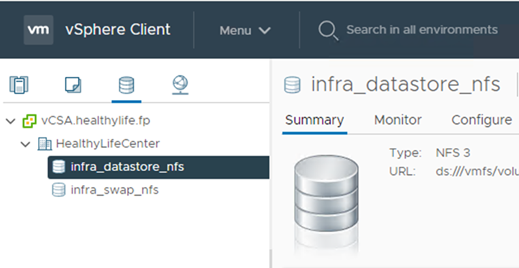
Adicione o datastore ao vCenter.
-
Usando o vCenter, adicione um datastore NFS aos hosts ESXi.
-
Usando o vCenter, crie uma VM Red Hat Enterprise Linux (RHEL) 8,3 para executar o GATK.
-
Um datastore NFS é apresentado à VM e montado no
/mnt/genomics, que é usado para armazenar executáveis GATK, scripts, arquivos de mapa de alinhamento binário (BAM), arquivos de referência, arquivos de índice, arquivos de dicionário e arquivos de saída para chamadas variantes.
Configuração e execução do GATK
Instale os seguintes pré-requisitos na VM Linux do RedHat Enterprise 8,3:
-
Java 8 ou SDK 1,8 ou posterior
-
Baixe o GATK 4.2.0.0 do Broad Institute "Site do GitHub". Os dados de sequência do genoma são geralmente armazenados na forma de uma série de colunas ASCII delimitadas por tabulações. No entanto, ASCII ocupa muito espaço para armazenar. Portanto, um novo padrão evoluiu chamado um arquivo BAM (.bam). Um arquivo BAM armazena os dados de sequência em uma forma compactada, indexada e binária. Nós "transferido" um conjunto de arquivos BAM disponíveis publicamente para execução do GATK a partir do "domínio público". Também baixamos arquivos de índice (.bai), arquivos de dicionário (. Dict) e arquivos de dados de referência (. fasta) do mesmo domínio público.
Após o download, o kit de ferramentas GATK tem um arquivo jar e um conjunto de scripts de suporte.
-
gatk-package-4.2.0.0-local.jarexecutável -
gatkficheiro de script.
Nós baixamos os arquivos BAM e os arquivos correspondentes de índice, dicionário e genoma de referência para uma família que consistia em arquivos pai, mãe e filho *.bam.
Motor Cromwell
Cromwell é um mecanismo de código aberto voltado para fluxos de trabalho científicos que permite o gerenciamento de fluxos de trabalho. O mecanismo Cromwell pode ser executado em dois "modos" modos , servidor ou em um modo de execução de fluxo de trabalho único. O comportamento do motor Cromwell pode ser controlado com o "Ficheiro de configuração do motor Cromwell".
-
Modo servidor. Permite "Repousante" a execução de fluxos de trabalho no motor Cromwell.
-
Modo de execução. O modo de execução é mais adequado para a execução de fluxos de trabalho individuais no Cromwell, "ref" para um conjunto completo de opções disponíveis no modo de execução.
Usamos o motor Cromwell para executar os fluxos de trabalho e pipelines em escala. O motor Cromwell usa uma linguagem de script baseada em WDL (user-friendly "idioma de descrição do fluxo de trabalho"). Cromwell também suporta um segundo padrão de script de fluxo de trabalho chamado linguagem de fluxo de trabalho comum (CWL). Ao longo deste relatório técnico, utilizamos a WDL. O WDL foi originalmente desenvolvido pelo Instituto amplo para pipelines de análise de genoma. O uso dos fluxos de trabalho WDL pode ser implementado usando várias estratégias, incluindo as seguintes:
-
* Encadeamento linear.* Como o nome sugere, a saída da tarefa nº 1 é enviada para a tarefa nº 2 como entrada.
-
* Multi-in / out. * Isso é semelhante ao encadeamento linear, na medida em que cada tarefa pode ter várias saídas sendo enviadas como entrada para tarefas subsequentes.
-
Scatter-GET. Essa é uma das estratégias de integração de aplicativos empresariais (EAI) mais poderosas disponíveis, especialmente quando usada em arquitetura orientada a eventos. Cada tarefa é executada de forma dissociada, e a saída para cada tarefa é consolidada na saída final.
Há três etapas quando o WDL é usado para executar o GATK em um modo autônomo:
-
Valide a sintaxe
womtool.jarusando .[root@genomics1 ~]# java -jar womtool.jar validate ghplo.wdl
-
Gerar entradas JSON.
[root@genomics1 ~]# java -jar womtool.jar inputs ghplo.wdl > ghplo.json
-
Execute o fluxo de trabalho usando o mecanismo Cromwell e
Cromwell.jar.[root@genomics1 ~]# java -jar cromwell.jar run ghplo.wdl –-inputs ghplo.json
O GATK pode ser executado usando vários métodos; este documento explora três desses métodos.
Execução do GATK usando o arquivo jar
Vamos olhar para uma única execução de pipeline de chamada variante usando o chamador da variante haplotype.
[root@genomics1 ~]# java -Dsamjdk.use_async_io_read_samtools=false \ -Dsamjdk.use_async_io_write_samtools=true \ -Dsamjdk.use_async_io_write_tribble=false \ -Dsamjdk.compression_level=2 \ -jar /mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar \ HaplotypeCaller \ --input /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ --output workshop_1906_2-germline_bams_father.validation.vcf \ --reference /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta
Neste método de execução, usamos o arquivo jar de execução local GATK, usamos um único comando java para invocar o arquivo jar, e passamos vários parâmetros para o comando.
-
Este parâmetro indica que estamos invocando o
HaplotypeCallerpipeline de chamadas variante. -
-- inputEspecifica o arquivo BAM de entrada. -
--outputespecifica o arquivo de saída da variante no formato de chamada variante (*.vcf("ref") ). -
Com o
--referenceparâmetro, estamos passando um genoma de referência.
Uma vez executado, os detalhes de saída podem ser encontrados na seção "Saída para execução do GATK usando o arquivo jar."
Execução do GATK usando script ./gatk
O kit de ferramentas GATK pode ser executado usando o ./gatk script. Vamos examinar o seguinte comando:
[root@genomics1 execution]# ./gatk \ --java-options "-Xmx4G" \ HaplotypeCaller \ -I /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ -R /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta \ -O /mnt/genomics/GATK/TEST\ DATA/variants.vcf
Passamos vários parâmetros para o comando.
-
Este parâmetro indica que estamos invocando o
HaplotypeCallerpipeline de chamadas variante. -
-IEspecifica o arquivo BAM de entrada. -
-Oespecifica o arquivo de saída da variante no formato de chamada variante (*.vcf("ref") ). -
Com o
-Rparâmetro, estamos passando um genoma de referência.
Uma vez executado, os detalhes de saída podem ser encontrados na seção
Execução do GATK usando o motor Cromwell
Usamos o motor Cromwell para gerenciar a execução do GATK. Vamos examinar a linha de comando e seus parâmetros.
[root@genomics1 genomics]# java -jar cromwell-65.jar \ run /mnt/genomics/GATK/seq/ghplo.wdl \ --inputs /mnt/genomics/GATK/seq/ghplo.json
Aqui, invocamos o comando Java passando o -jar parâmetro para indicar que pretendemos executar um arquivo jar, por exemplo, Cromwell-65.jar. O próximo parâmetro passado (run) indica que o motor Cromwell está em funcionamento no modo Run (Executar), a outra opção possível é o modo Server (servidor). O próximo parâmetro é *.wdl que o modo Run deve ser usado para executar os pipelines. O próximo parâmetro é o conjunto de parâmetros de entrada para os fluxos de trabalho que estão sendo executados.
Veja como é o conteúdo do ghplo.wdl arquivo:
[root@genomics1 seq]# cat ghplo.wdl
workflow helloHaplotypeCaller {
call haplotypeCaller
}
task haplotypeCaller {
File GATK
File RefFasta
File RefIndex
File RefDict
String sampleName
File inputBAM
File bamIndex
command {
java -jar ${GATK} \
HaplotypeCaller \
-R ${RefFasta} \
-I ${inputBAM} \
-O ${sampleName}.raw.indels.snps.vcf
}
output {
File rawVCF = "${sampleName}.raw.indels.snps.vcf"
}
}
[root@genomics1 seq]#
Aqui está o arquivo JSON correspondente com as entradas para o motor Cromwell.
[root@genomics1 seq]# cat ghplo.json
{
"helloHaplotypeCaller.haplotypeCaller.GATK": "/mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar",
"helloHaplotypeCaller.haplotypeCaller.RefFasta": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta",
"helloHaplotypeCaller.haplotypeCaller.RefIndex": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta.fai",
"helloHaplotypeCaller.haplotypeCaller.RefDict": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.dict",
"helloHaplotypeCaller.haplotypeCaller.sampleName": "fatherbam",
"helloHaplotypeCaller.haplotypeCaller.inputBAM": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bam",
"helloHaplotypeCaller.haplotypeCaller.bamIndex": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bai"
}
[root@genomics1 seq]#
Por favor, note que Cromwell usa um banco de dados na memória para a execução. Uma vez executado, o log de saída pode ser visto na seção "Saída para execução do GATK usando o motor Cromwell."
Para obter um conjunto abrangente de etapas sobre como executar o GATK, consulte o "Documentação do GATK".