

Genomics - GATK setup and execution
 Suggest changes
Suggest changes
According to the National Human Genome Research Institute ( NHGRI), “Genomics is the study of all of a person’s genes (the genome), including interactions of these genes with each other and with a person’s environment. ”
According to the NHGRI, “Deoxyribonucleic acid (DNA) is the chemical compound that contains the instructions needed to develop and direct the activities of nearly all living organisms. DNA molecules are made of two twisting, paired strands, often referred to as a double helix.” “An organism’s complete set of DNA is called its genome.”
Sequencing is the process of determining the exact order of the bases in a strand of DNA. One of the most common types of sequencing used today is called sequencing by synthesis. This technique uses the emission of fluorescent signals to order the bases. Researchers can use DNA sequencing to search for genetic variations and any mutations that might play a role in the development or progression of a disease while a person is still in the embryonic stage.
From sample to variant identification, annotation, and prediction
At a high level, genomics can be classified into the following steps. This is not an exhaustive list:
-
Sample collection.
-
Genome sequencing using a sequencer to generate the raw data.
-
Preprocessing. For example, deduplication using Picard.
-
Genomic analysis.
-
Mapping to a reference genome.
-
Variant identification and annotation typically performed using GATK and similar tools.
-
-
Integration into the electronic health record (EHR) system.
-
Population stratification and identification of genetic variation across geographical location and ethnic background.
-
Predictive models using significant single- nucleotide polymorphism.
The following figure shows the process from sampling to variant identification, annotation, and prediction.
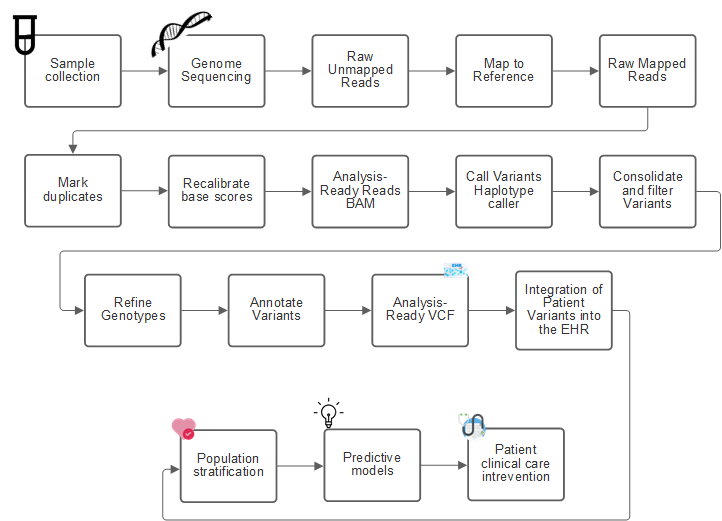
The Human Genome project was completed in April 2003, and the project made a very high-quality simulation of the human genome sequence available in the public domain. This reference genome initiated an explosion in research and development of genomics capabilities. Virtually every human ailment has a signature in that human’s genes. Up until recently, physicians were leveraging genes to predict and determine birth defects like sickle cell anemia, which is caused by a certain inheritance pattern caused by a change in a single gene. The treasure trove of data being made available by the human genome project led to the advent of the current state of genomics capabilities.
Genomics has a broad set of benefits. Here is a small set of benefits in the healthcare and life sciences domains:
-
Better diagnosis at point of care
-
Better prognosis
-
Precision medicine
-
Personalized treatment plans
-
Better disease monitoring
-
Reduction in adverse events
-
Improved access to therapies
-
Improved disease monitoring
-
Effective clinical trial participation and better selection of patients for clinical trials based on genotypes.
Genomics is a "four-headed beast," because of the computational demands across the lifecycle of a dataset: acquisition, storage, distribution, and analysis.
Genome Analysis Toolkit (GATK)
GATK was developed as a data science platform at the Broad Institute. GATK is a set of open-source tools that enable genome analysis, specifically variant discovery, identification, annotation, and genotyping. One of the benefits of GATK is that the set of tools and or commands can be chained to form a complete workflow. The primary challenges that Broad institute tackles are the following:
-
Understand the root causes and biological mechanisms of diseases.
-
Identify therapeutic interventions that act at the fundamental cause of a disease.
-
Understand the line of sight from variants to function in human physiology.
-
Create standards and policy frameworks for genome data representation, storage, analysis, security, and so on.
-
Standardize and socialize interoperable genome aggregation databases (gnomAD).
-
Genome-based monitoring, diagnosis, and treatment of patients with greater precision.
-
Help implement tools that predict diseases well before symptoms appear.
-
Create and empower a community of cross-disciplinary collaborators to help tackle the toughest and most important problems in biomedicine.
According to GATK and the Broad institute, genome sequencing should be treated as a protocol in a pathology lab; every task is well documented, optimized, reproducible, and consistent across samples and experiments. The following is a set of steps recommended by the Broad Institute, for more information, see the GATK website.
FlexPod setup
Genomics workload validation includes a from-scratch setup of a FlexPod infrastructure platform. The FlexPod platform is highly available and can be independently scaled; for example, network, storage and compute can be scaled independently. We used the following Cisco validated design guide as the reference architecture document to set up the FlexPod environment: FlexPod Datacenter with VMware vSphere 7.0 and NetApp ONTAP 9.7. See the following FlexPod platform set up highlights:
To perform FlexPod lab setup, complete the following steps:
-
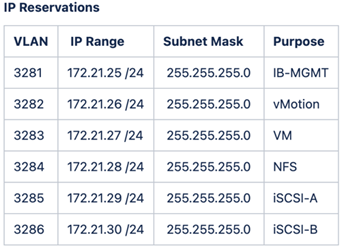
FlexPod lab setup and validation uses the following IP4 reservations and VLANs.
-
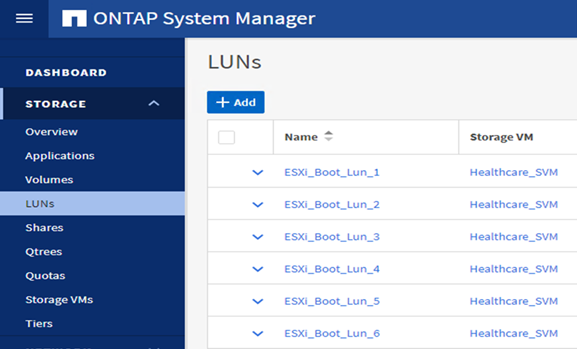
Configure iSCSI-based boot LUNs on the ONTAP SVM.
-
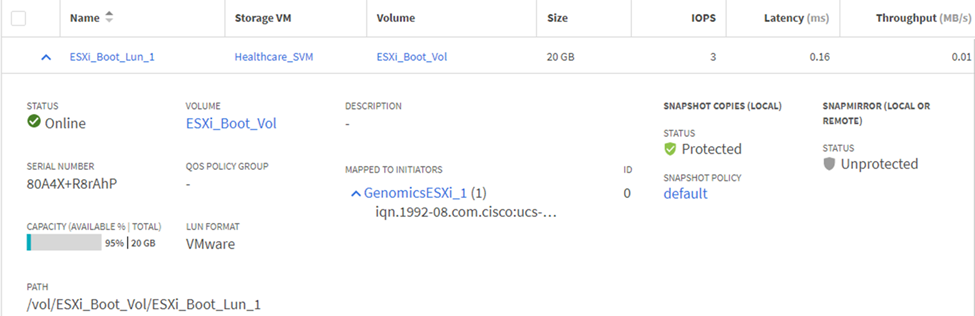
Map LUNs to iSCSI initiator groups.
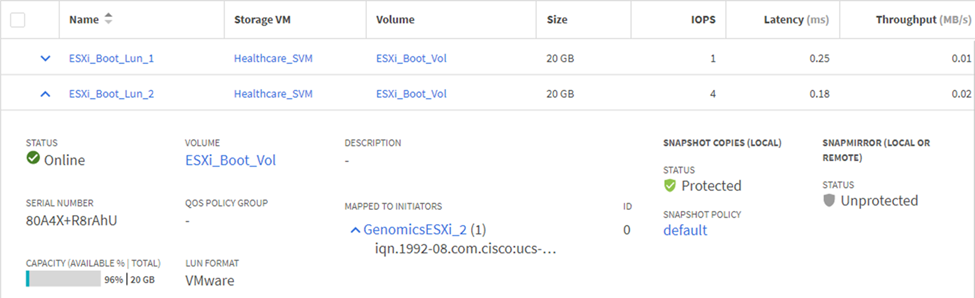
-
Install vSphere 7.0 with iSCSI boot.
-
Register ESXi hosts with the vCenter.
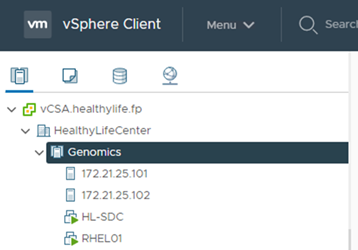
-
Provision an NFS datastore
infra_datastore_nfson the ONTAP storage.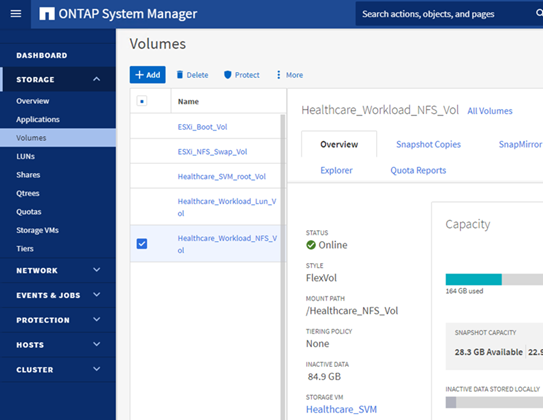
-
Add the datastore to the vCenter.
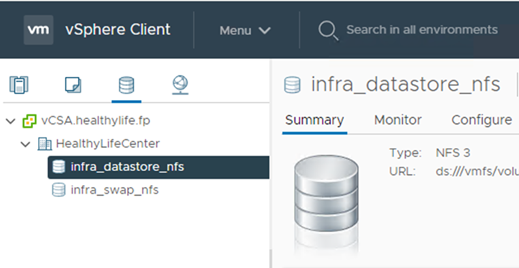
-
Using vCenter, add an NFS datastore to the ESXi hosts.
-
Using the vCenter, create a Red Hat Enterprise Linux (RHEL) 8.3 VM to run GATK.
-
An NFS datastore is presented to the VM and mounted at
/mnt/genomics, which is used to store GATK executables, scripts, Binary Alignment Map (BAM) files, reference files, index files, dictionary files, and out files for variant calling.
GATK setup and execution
Install the following prerequisites on the RedHat Enterprise 8.3 Linux VM:
-
Java 8 or SDK 1.8 or later
-
Download GATK 4.2.0.0 from the Broad Institute GitHub site. Genome sequence data is generally stored in the form of a series of tab-delimited ASCII columns. However, ASCII takes too much space to store. Therefore, a new standard evolved called a BAM (*.bam) file. A BAM file stores the sequence data in a compressed, indexed, and binary form. We downloaded a set of publicly available BAM files for GATK execution from the public domain. We also downloaded index files (\*.bai), dictionary files (*. dict), and reference data files (*. fasta) from the same public domain.
After downloading, the GATK tool kit has a jar file and a set of support scripts.
-
gatk-package-4.2.0.0-local.jarexecutable -
gatkscript file.
We downloaded the BAM files and the corresponding index, dictionary, and reference genome files for a family that consisted of father, mother, and son *.bam files.
Cromwell engine
Cromwell is an open-source engine geared towards scientific workflows that enables workflow management. The Cromwell engine can be run in two modes, Server mode or a single- workflow Run mode. The behavior of the Cromwell engine can be controlled using the Cromwell engine configuration file.
We use the Cromwell engine to execute the workflows and pipelines at scale. The Cromwell engine uses a user-friendly workflow description language (WDL)-based scripting language. Cromwell also supports a second workflow scripting standard called the common workflow language (CWL). Throughout this technical report, we used WDL. WDL was originally developed by the Broad Institute for genome analysis pipelines. Using the WDL workflows can be implemented using several strategies, including the following:
-
Linear chaining. As the name suggests, output from task#1 is sent to task #2 as input.
-
Multi-in/out. This is similar to linear chaining in that each task can have multiple outputs being sent as input to subsequent tasks.
-
Scatter-gather. This is one of the most powerful enterprise application integration (EAI) strategies available, especially when used in event-driven architecture. Each task executes in a decoupled fashion, and the output for each task is consolidated into the final output.
There are three steps when WDL is used to run GATK in a standalone mode:
-
Validate syntax using
womtool.jar.[root@genomics1 ~]# java -jar womtool.jar validate ghplo.wdl
-
Generate inputs JSON.
[root@genomics1 ~]# java -jar womtool.jar inputs ghplo.wdl > ghplo.json
-
Run the workflow using the Cromwell engine and
Cromwell.jar.[root@genomics1 ~]# java -jar cromwell.jar run ghplo.wdl –-inputs ghplo.json
The GATK can be executed by using several methods; this document explores three of these methods.
Execution of GATK using the jar file
Let’s look at a single variant call pipeline execution using the Haplotype variant caller.
[root@genomics1 ~]# java -Dsamjdk.use_async_io_read_samtools=false \ -Dsamjdk.use_async_io_write_samtools=true \ -Dsamjdk.use_async_io_write_tribble=false \ -Dsamjdk.compression_level=2 \ -jar /mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar \ HaplotypeCaller \ --input /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ --output workshop_1906_2-germline_bams_father.validation.vcf \ --reference /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta
In this method of execution, we use the GATK local execution jar file, we use a single java command to invoke the jar file, and we pass several parameters to the command.
-
This parameter indicates that we are invoking the
HaplotypeCallervariant caller pipeline. -
-- inputspecifies the input BAM file. -
--outputspecifies the variant output file in variant call format (*.vcf) (ref). -
With the
--referenceparameter, we are passing a reference genome.
Once executed, output details can be found in the section "Output for execution of GATK using the jar file."
Execution of GATK using ./gatk script
GATK tool kit can be executed using the ./gatk script. Let’s examine the following command:
[root@genomics1 execution]# ./gatk \ --java-options "-Xmx4G" \ HaplotypeCaller \ -I /mnt/genomics/GATK/TEST\ DATA/bam/workshop_1906_2-germline_bams_father.bam \ -R /mnt/genomics/GATK/TEST\ DATA/ref/workshop_1906_2-germline_ref_ref.fasta \ -O /mnt/genomics/GATK/TEST\ DATA/variants.vcf
We pass several parameters to the command.
-
This parameter indicates that we are invoking the
HaplotypeCallervariant caller pipeline. -
-Ispecifies the input BAM file. -
-Ospecifies the variant output file in variant call format (*.vcf) (ref). -
With the
-Rparameter, we are passing a reference genome.
Once executed, output details can be found in the section "Output for execution of GATK using the ./gatk script."
Execution of GATK using Cromwell engine
We use the Cromwell engine to manage GATK execution. Let’s examine the command line and it’s parameters.
[root@genomics1 genomics]# java -jar cromwell-65.jar \ run /mnt/genomics/GATK/seq/ghplo.wdl \ --inputs /mnt/genomics/GATK/seq/ghplo.json
Here, we invoke the Java command by passing the -jar parameter to indicate that we intend to execute a jar file, for example, Cromwell-65.jar. The next parameter passed (run) indicates that the Cromwell engine is running in Run mode, the other possible option is Server mode. The next parameter is *.wdl that the Run mode should use to execute the pipelines. The next parameter is the set of input parameters to the workflows being executed.
Here’s what the contents of the ghplo.wdl file look like:
[root@genomics1 seq]# cat ghplo.wdl
workflow helloHaplotypeCaller {
call haplotypeCaller
}
task haplotypeCaller {
File GATK
File RefFasta
File RefIndex
File RefDict
String sampleName
File inputBAM
File bamIndex
command {
java -jar ${GATK} \
HaplotypeCaller \
-R ${RefFasta} \
-I ${inputBAM} \
-O ${sampleName}.raw.indels.snps.vcf
}
output {
File rawVCF = "${sampleName}.raw.indels.snps.vcf"
}
}
[root@genomics1 seq]#
Here’s the corresponding JSON file with the inputs to the Cromwell engine.
[root@genomics1 seq]# cat ghplo.json
{
"helloHaplotypeCaller.haplotypeCaller.GATK": "/mnt/genomics/GATK/gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar",
"helloHaplotypeCaller.haplotypeCaller.RefFasta": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta",
"helloHaplotypeCaller.haplotypeCaller.RefIndex": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.fasta.fai",
"helloHaplotypeCaller.haplotypeCaller.RefDict": "/mnt/genomics/GATK/TEST DATA/ref/workshop_1906_2-germline_ref_ref.dict",
"helloHaplotypeCaller.haplotypeCaller.sampleName": "fatherbam",
"helloHaplotypeCaller.haplotypeCaller.inputBAM": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bam",
"helloHaplotypeCaller.haplotypeCaller.bamIndex": "/mnt/genomics/GATK/TEST DATA/bam/workshop_1906_2-germline_bams_father.bai"
}
[root@genomics1 seq]#
Please note that Cromwell uses an in-memory database for the execution. Once executed, the output log can be seen in the section "Output for execution of GATK using the Cromwell engine."
For a comprehensive set of steps on how to execute GATK, see the GATK documentation.